T-cell stimulating vaccines empower CD3 bispecific antibody therapy in solid tumors
Published in Cancer
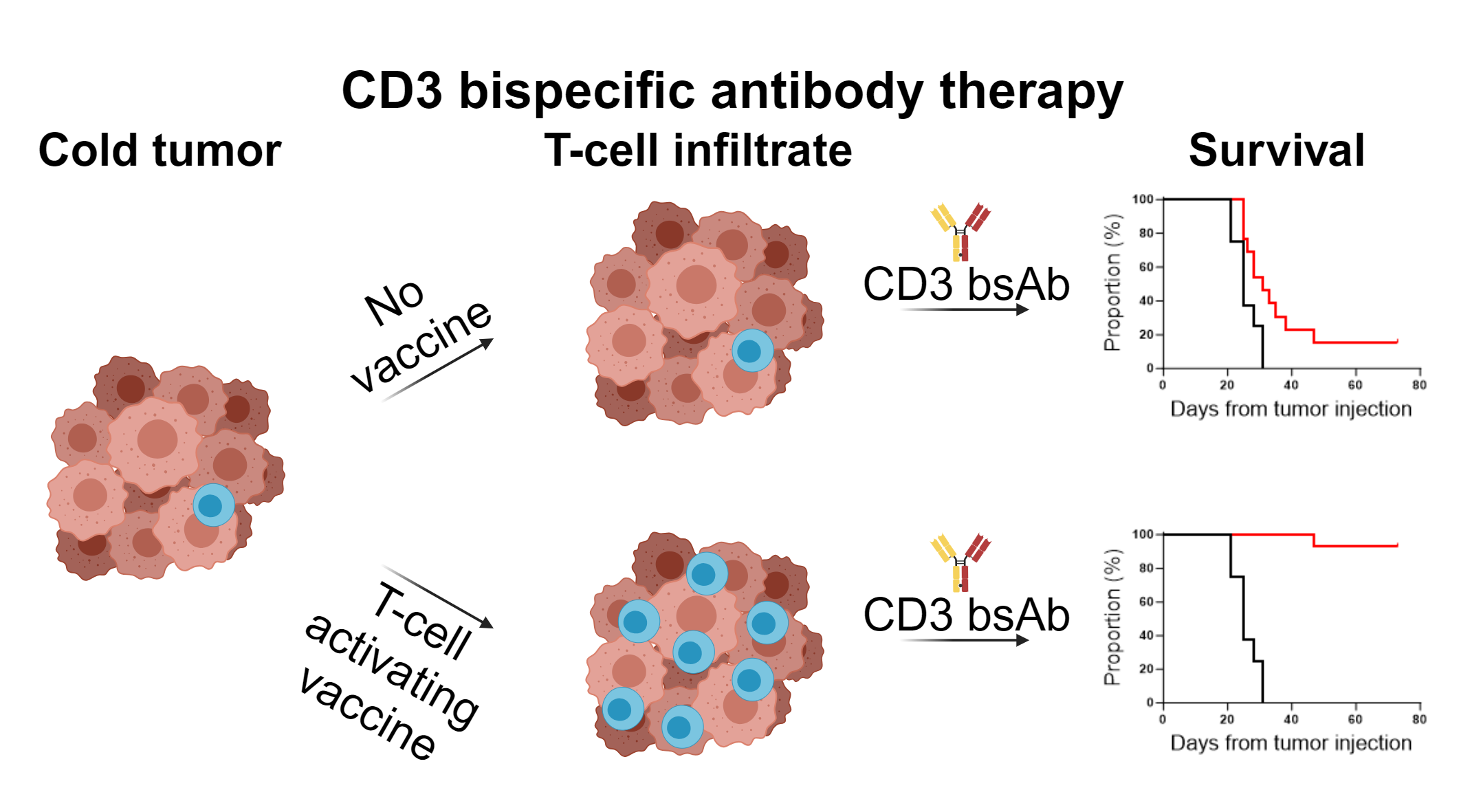
Background
Tumors do not only exist of transformed cells, but also comprise infiltrating immune cells, such as macrophages and T cells1. Local interactions between tumor cells and immune cells are intricate and complex with for example killer T cells attempting to eradicate the tumor. However, in the case of clinically evident cancers, these attempts were unsuccessful. Immunotherapy aims to help the immune cells to successfully clear the tumor. Over the last decade CD3 bispecific antibody (CD3 bsAb) or T-cell engager therapy established itself as a potent off-the-shelf immunotherapeutic strategy in the treatment arsenal of oncologists. These CD3 bsAbs activate killer T cells by cross-linking the T cells with a tumor protein on the surface of tumor cells, resulting in the formation of an immunological synapse and subsequent tumor cell killing2. An important aspect of this therapy is that the engagement of T cells via CD3ε is independent of the specificity of their T-cell receptor (TCR), thereby enabling all (bystander) T cells in the tumor as potent anti-tumor effector cells3. As one can imagine, this type of therapy works particularly well for hematological malignancies, where tumor cells are surrounded by T cells and are easily accessible for the bsAb. Indeed, five different CD3 bsAbs have been approved for hematological cancers, with all of them targeting B-cell antigens, in contrast to solid tumors, where tebentafusp is the only CD3 bsAb that has been approved for the treatment of advanced uveal melanoma4,5. So, solid cancers represent a bigger challenge due to additional hurdles, such as low levels of T-cell infiltration and a hostile tumor microenvironment (TME) caused by the low pH, hypoxia, nutrient deprivation and high concentration of oxygen radicals, all of which can hamper T-cell effector functions6.
In our manuscript we aimed to overcome the hurdles of poor T-cell infiltrate and the hypofunctionality of existing tumor-resident T cells via the administration of T-cell activating vaccines before application of CD3 bsAb therapy.
Key findings
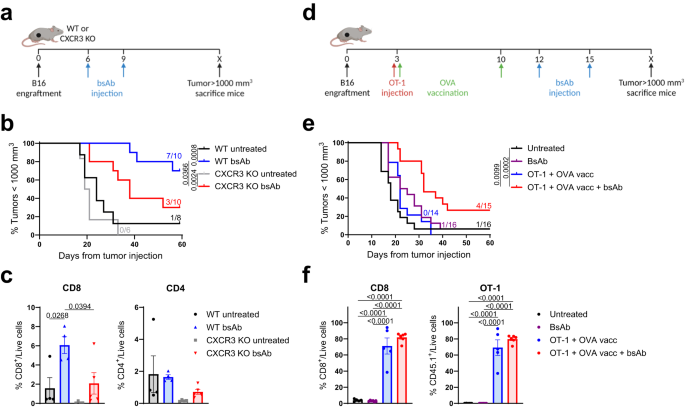
We first investigated the dependency of T-cell influx in our model using CXCR3 KO mice, lacking this key receptor for T-cell trafficking towards tumors, and found that CD3xTRP1 treatment efficacy was impaired in these mice. As this demonstrated that T-cell influx is important for the therapeutic outcome, we hypothesized that increasing T-cell infiltration could potentially improve the efficacy of CD3 bsAb therapy. We increased T-cell infiltration by adoptive transfer of tumor-nonspecific OT-1 CD8 T cells in combination with synthetic long peptide vaccination encompassing the OVA epitope supplemented with imiquimod as adjuvant and IL-2. Indeed, we found that OT-1 transfer and OVA vaccination generated profound T-cell infiltration in the tumor, and more importantly, that subsequent CD3xTRP1 administration resulted in superior survival over bsAb monotherapy. We then moved on to study the kinetics of the OT-1 T cells for this triple combination using in vivo imaging (IVIS) approaches and genetically engineered OT-1 T cells, allowing us to separately trace T-cell presence and T-cell activation in mice bearing TRP1-positive or -negative tumors in distinct flanks. We observed that OT-1 transfer and OVA vaccination generated systemic influx of OT-1 T cells in both the TRP1-positive and -negative tumors despite the fact that neither of them expressed the OVA antigen. Subsequent CD3xTRP1 administration resulted in activation of the OT-1 T cells specifically in TRP1-positive tumors.
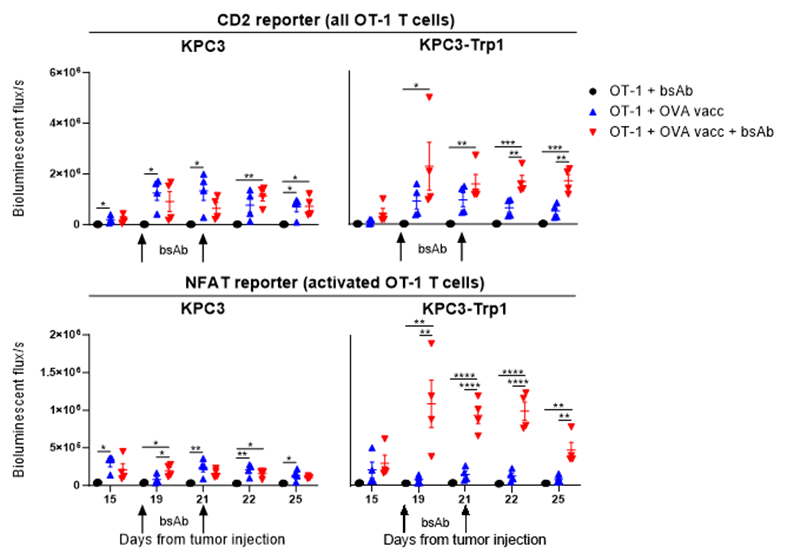
Figure 1: In vivo imaging of OT-1 T cells in mice bearing KPC3 and KPC3-TRP1 tumors treated with OT-1 transfer, OVA vaccination and CD3xTRP1 bsAb.
We then performed in depth analysis of the TME for the triple combination treatment, including transcriptomics, high dimensional flow cytometry and immunohistochemistry and confirmed that 1) OT-1 transfer and OVA vaccination generated intratumoral T-cell influx and 2) subsequent CD3xTRP1 administration resulted in local T-cell activation in antigen-positive tumors and more profound infiltration depth of the T cells. Apart from an enormous OT-1 influx, we also detected significant frequencies of endogenous OVA-specific T cells in tumors due to OVA vaccination. We then studied the importance of these endogenous cells for the combination therapy by again using CXCR3 KO mice and transferring WT OT-1 T cells, thereby only impairing the influx of endogenous T cells, and found that the endogenous T cells were crucial for durable treatment responses despite the abundance of OT-1 T cells. Therefore, we decided to move away from the artificial OT-1 transfer and only use diverse vaccination approaches to induce endogenous T-cell responses. An extensive TME analysis revealed that combination of OVA vaccination and CD3xTRP1 resulted in extensive pro-inflammatory remodeling of the TME, increasing numbers of activated T cells, NK cells and pro-inflammatory myeloid cells. Finally, we showed that these profound changes in the TME translated into better anti-tumor activity and survival, as combination with multiple vaccination strategies including synthetic long peptides and live viral infections improved CD3xTRP1 treatment efficacy.
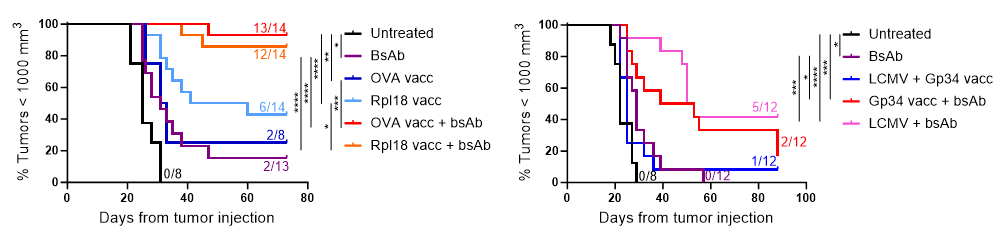
Figure 2: Kaplan-Meier graphs of mice bearing MC38-TRP1 tumors (left) or B16F10 tumors (right) treated with tumor-nonspecific vaccination (synthetic long peptide: OVA and Gp34 from LCMV; infection: LCMV) or tumor-specific vaccination (synthetic long peptide: Rpl18) in combination with CD3xTRP1 bsAb.
Take home message
Efficacy of CD3 bispecific antibody therapy faces severe challenges in solid tumors, including poor T-cell infiltration and an immunosuppressive TME. Here, we provide a strategy to overcome these hurdles by pre-conditioning CD3 bsAb with vaccination approaches. We demonstrate that application of both tumor-specific and non-specific vaccination prior to CD3 bsAb administration enhances T-cell infiltration, pro-inflammatory TME polarization and improves treatment responses in immunologically ‘cold’ tumors. We believe that combining CD3 bsAbs together with vaccine modalities capable of eliciting potent T-cell responses could soon lead to a successful clinical translation of this combination therapy for solid tumor indications.
References
- Fridman, W. H., Pagès, F., Sautès-Fridman, C. & Galon, J. The immune contexture in human tumours: impact on clinical outcome. Nature reviews. Cancer vol. 12 298–306 (2012).
- Wei, J., Yang, Y., Wang, G. & Liu, M. Current landscape and future directions of bispecific antibodies in cancer immunotherapy. Front. Immunol. 13, 1035276 (2022).
- Baeuerle, P. A. & Wesche, H. T-cell-engaging antibodies for the treatment of solid tumors: challenges and opportunities. Curr. Opin. Oncol. 34, 552–558 (2022).
- van de Donk, N. W. C. J. & Zweegman, S. T-cell-engaging bispecific antibodies in cancer. Lancet (London, England) 402, 142–158 (2023).
- Nathan, P. et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 385, 1196–1206 (2021).
- Middelburg, J. et al. Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors. Cancers (Basel). 13, 1–25 (2021).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in