Taking Synaptic Vesicle Proteins Out of the Membrane and Into the Light
Published in Ecology & Evolution, Neuroscience, and General & Internal Medicine
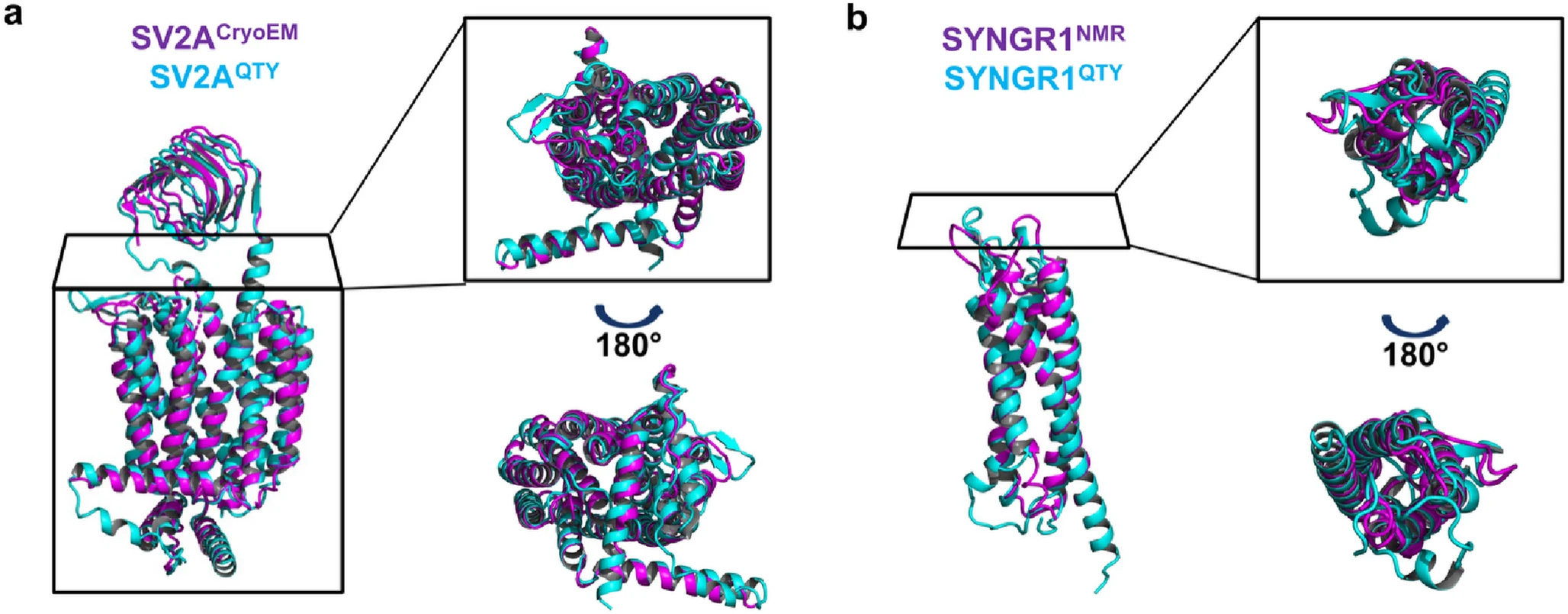
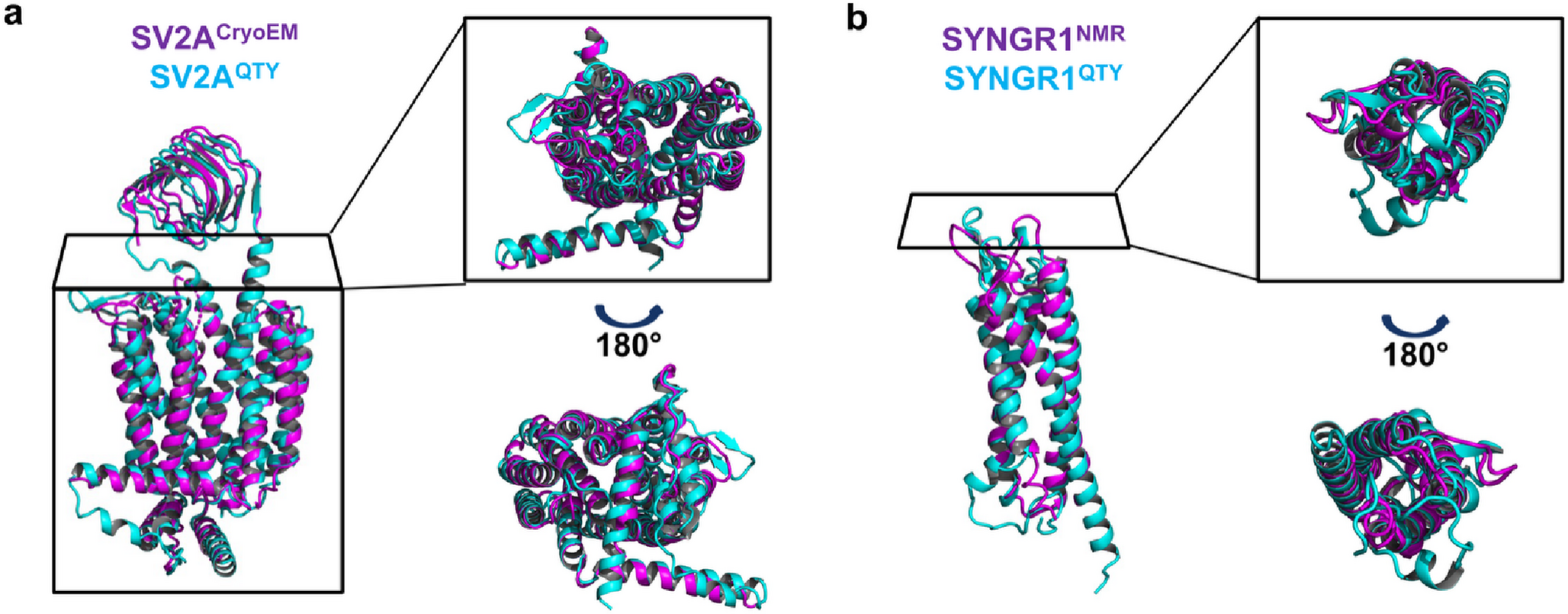
Our team asked a simple but ambitious question: What if we could redesign these integral membrane proteins to be completely water-soluble without destroying their 3D architecture? Here is a look behind the scenes of our recent paper, where we used the QTY Code alongside molecular dynamics and evolutionary bioinformatics to untangle the mysteries of synaptic vesicle proteins.
The QTY Code: A Molecular Trick
To pull these proteins out of the lipid bilayer and into an aqueous environment, we employed the QTY code (Karagöl et al. 2024a, 2024b,2025a,2025b,2025c). This approach systematically replaces specific hydrophobic amino acids with hydrophilic ones that have similar side-chain volumes and electron densities. Specifically, we substituted:
- Leucine (L) with Glutamine (Q)
- Valine/Isoleucine (V/I) with Threonine (T)
- Phenylalanine (F) with Tyrosine (Y)
When we applied this code to eight synaptic vesicle proteins, it required changing up to 55% of their transmembrane sequences. Changing half of a protein's core sequence would usually guarantee that it misfolds or collapses. Remarkably, our newly designed water-soluble variants maintained incredible structural similarity to their native counterparts, with root mean square deviation (RMSD) values remaining below 1.9Å.
Validating the Structures
We didn't just want to rely on static predictions. To ensure these new variants were dynamically stable, we ran 100-nanosecond molecular dynamics simulations. The results were thrilling: the QTY substitutions preserved the overall fold integrity of the proteins. The core transmembrane residues exhibited minimal atomic-level disturbances, proving that QTY helices can successfully emulate the dynamic stability of native transmembrane helices even when floating freely in water.
Looking at Nature: We Aren't the Only Ones Using QTY
While QTY was developed as a synthetic engineering tool, we wondered if nature utilizes the same structural trick. We scoured human genomic databases and found 155 natural single nucleotide variants (SNVs) that perfectly mirror QTY (L to Q, I to T, F to Y) or reverse-QTY substitutions within these synaptic proteins.
When we evaluated these natural variants using the AI-powered AlphaMissense tool, we uncovered a striking evolutionary pattern: QTY-like variants were significantly more likely to be predicted as benign compared to other random polar or non-polar missense mutations. This suggests that evolution inherently tolerates these specific isostructural polarity shifts because they conserve side-chain volume and geometry, overriding the energetic costs normally associated with swapping a hydrophobic residue for a hydrophilic one.
The Threonine-Valine Mystery
Perhaps the most surprising finding of our evolutionary analysis was the strong evolutionary relationship between Threonine (T) and Valine (V) frequencies in homologous sequences.
This coupling is fascinating because substituting Threonine for Valine requires a double-nucleotide change in a single codon—an event with an incredibly high mutational barrier. How did nature bypass this barrier? By mapping these evolutionary trajectories using ancestral sequence reconstruction, our data indicated an "Alanine-centric" evolutionary model. Rather than direct leaps from T to V, Alanine likely served as a permissive functional intermediate, allowing parallel evolutionary pathways (A to V and A to T) to occur over time.
Why This Matters
Ultimately, these water-soluble QTY variants provide validated, highly stable models that bypass the immense challenges of membrane protein crystallography and purification.
Beyond structural biology, these proteins can serve as water-soluble decoys, offering a fresh foundation for drug discovery and innovative therapeutic strategies for neurological conditions related to synaptic dysfunction. By peering into both the synthetic potential and evolutionary past of these proteins, we now have a much clearer map of how proteins adapt to fundamentally different environments.
Follow the Topic
-
Journal of Molecular Evolution
This journal covers experimental, computational, and theoretical work aimed at deciphering features of molecular evolution and the processes bearing on these features.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in