What Do Membrane Proteins Really Want?
Published in Cancer, Chemistry, and Ecology & Evolution
Our fascination with membrane proteins started with a very practical challenge: how do you study them if they are so difficult to solubilize and crystallize? Hydrophobic α-helices bury themselves in membranes, making structural and biochemical work notoriously hard. In our earlier work on the QTY code, we asked a simple but bold question: Can we swap hydrophobic residues (L, I, V, F) with polar ones (Q, T, Y) in neurological transporters to make them soluble, without breaking their helical structure?
To our surprise, the answer was yes (Zhang et al. PNAS 2018; Karagöl et al. Pharm Res 2024; Karagöl et al. PLOS One 2024a; Johnsson et al. 2025). These substitutions, though chemically distinct, preserved fold and even function. It was a reminder that evolution tolerates more flexibility than textbooks suggest. But this raised deeper questions. If QTY substitutions work well in the lab, could evolution itself be quietly using similar rules? Do natural proteins already rely on such hydrophobic–polar swaps to diversify their functions, adapt to new environments, or buffer against deleterious mutations? These questions are not just academic. The answers shape function. Substitutions that evolution tolerates can preserve essential folds, maintain stability in different membrane environments, such as drug resistance. In contrast, catastrophic substitutions frequently underlie disease-causing variants or disrupt critical protein–protein interactions, contributing to conditions ranging from cancer to neurodegeneration.
Answering these questions required moving beyond biochemistry into quantitative modeling. To chase these questions, we zoomed out; from a handful of transporters to 504 unique human transmembrane proteins with millions of possible variants. We asked: Is there a hidden logic to residue exchange across the membrane proteome?

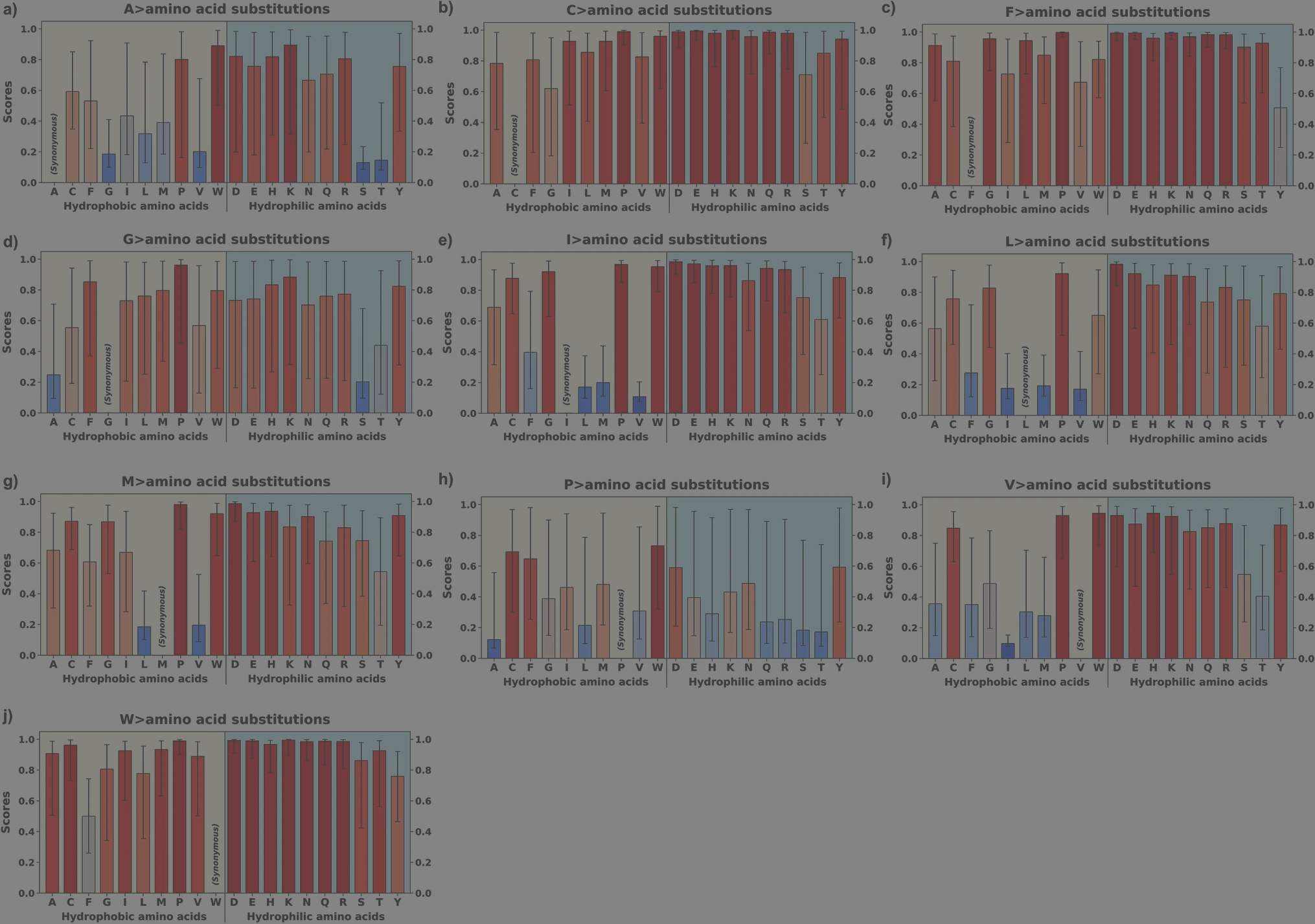
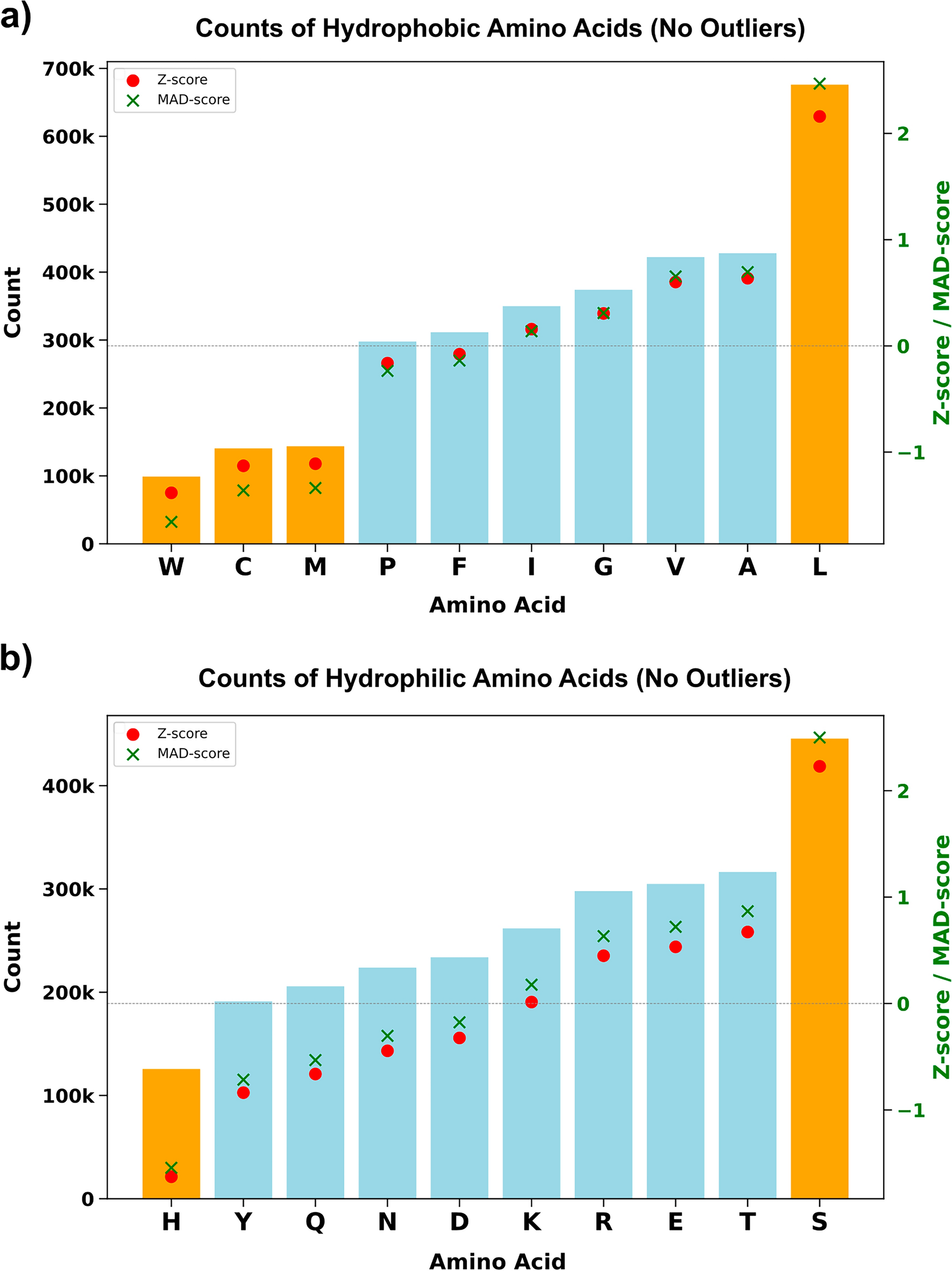
AlphaMissense pathogenicity scores of the variants from hydrophobic amino acids of structurally known alpha-helical transmembrane proteins. AlphaMissense scores, derived from machine learning algorithms trained on genomic and structural protein data, estimate the likelihood of amino acid substitutions being pathogenic to protein function. (DOI: 10.1007/s00239-025-10262-8)
The answers were surprising.
- Some substitutions we expected to be harmful, like F ↔ Y, turned out to be strikingly well-tolerated. Why does adding a single hydroxyl group to phenylalanine so rarely destabilize?
- Others showed clear asymmetry. An A > D substitution is often pathogenic, but D > A is relatively safe. Why is evolution kinder in one direction than the other?
- And then there was alanine. The unassuming residue emerged as an evolutionary hub - a bridge that allows otherwise improbable swaps like V ↔ T or G ↔ N through stepwise intermediates. Does alanine act as a molecular “negotiator” making the impossible possible?
To make sense of this, we turned to evolutionary game theory.
Game theory is a mathematical framework originally developed in economics to understand how rational “players” make decisions when their outcomes depend on each other. We here adapted it to biology - not because proteins or organisms consciously make choices, but because the same mathematical rules can describe their interactions.
What if substitutions are moves in a game, with residues competing or cooperating for stability and function? Suddenly, concepts like stochastic Nash equilibria made sense in the context of protein evolution.
- Evolutionarily Stable Strategies (ESS): Some substitutions become so robust that they dominate across evolution.
- Extensive forms: Some substitutions don’t happen directly but through stepwise paths. These are like decision trees in game theory, where intermediates open routes that would otherwise be blocked.
- Prisoner’s Dilemma: Alanine’s role is perhaps best understood here. Imagine residues at a site to stay in alanine or switch to V or T. Cooperation (staying in A) keeps options open, preserving flexibility. But there’s constant temptation to defect (to V or T) for immediate stability or hydrophobicity. Over time, this repeated game explains why alanine persists as a flexible hub, but also why polar-nonpolar asymmetries emerge in evolution.
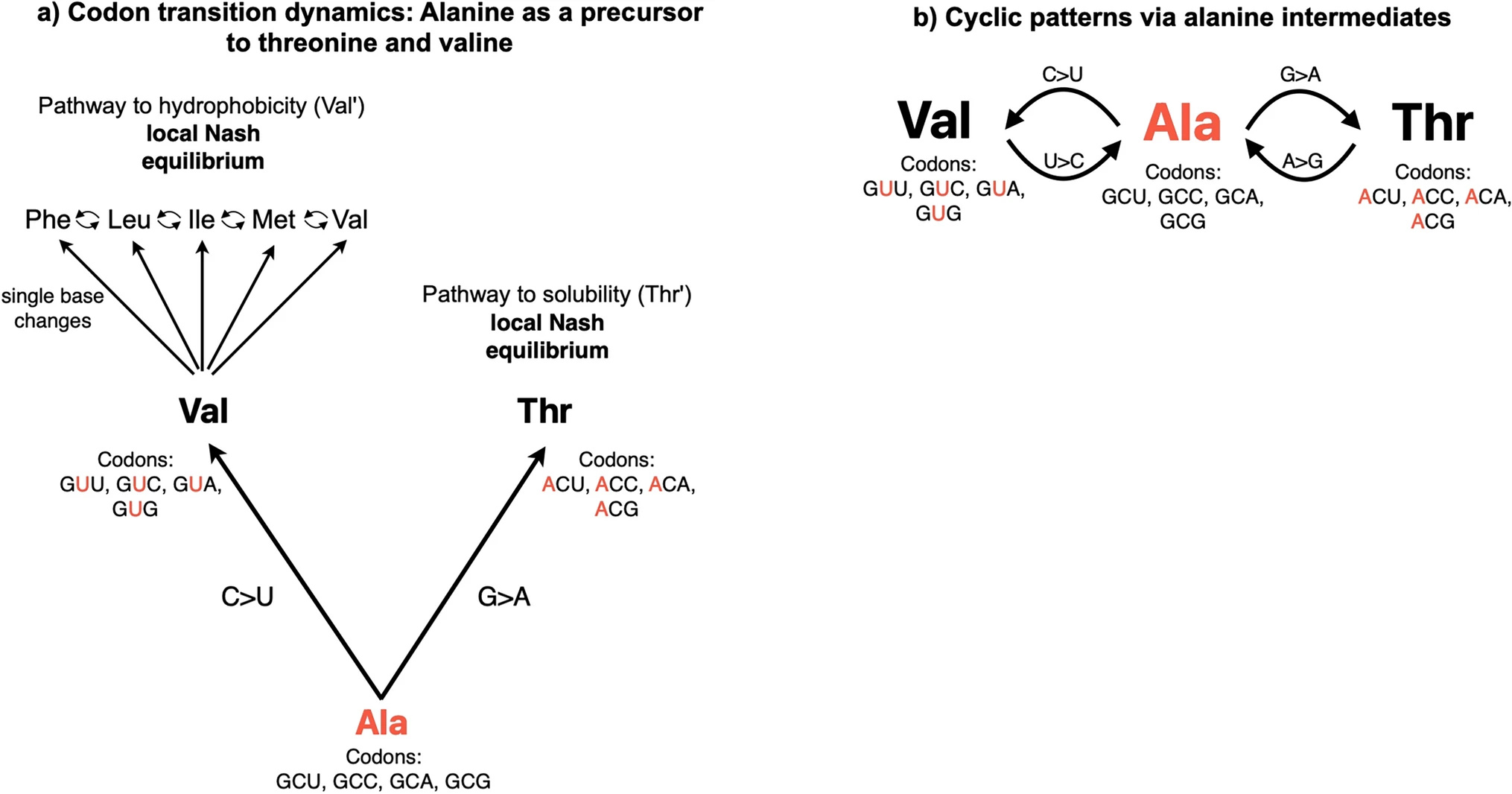
Alanine-centric model for the evolution of T-V relationship: This panel shows two evolutionary pathways centered around alanine (Ala). The "Pathway to hydrophobicity" (A > V) represents the evolution from alanine to valine and other nonpolar amino acids (F, L, I, M, V) through single base changes. The "Pathway to Solubility" shows the evolution from alanine to threonine (Thr). Both pathways are described as reaching a local Nash equilibrium. (DOI: 10.1007/s00239-025-10262-8)
But analogies are not enough; we needed to test whether these models held up in real evolutionary data. For this, we drew on several mathematical tools: Non-parametric correlation tests to capture nonlinear relationships between residue frequencies. Partial correlation analysis to control for conservation, teasing apart genuine co-evolution from background biases (Karagöl et al. PLOS One 2024b). Bayesian frameworks to weight evolutionary conservation across homologs. We further incorporated Hydropathy and charge analyses to test whether biochemical constraints aligned with the predicted game dynamics (Karagöl A & Karagöl T 2025).
The results were striking
When we controlled for conservation, positive correlations between alanine and both valine and threonine collapsed, exactly as predicted by the Prisoner’s Dilemma model: Alanine (A) acts as a hub, but once either V or T “defects” the correlation drops. Similarly, F ↔ Y substitutions formed a strong positive correlation, confirming what our QTY work had hinted at years earlier. Patterns were not symmetric. A > D could be harmful, while D > A was often tolerated. Some substitutions clustered into unexpected alliances. As a result, our study suggests a previously unrecognized evolutionary dynamic, potentially linked to functional diversification, which could be targeted to combat drug resistance and cancer.
In other words, the mathematics matched the biology. However, this left us with more questions than answers.
- Are these substitution “games” universal across all organisms, or do they shift under different evolutionary pressures?
- Could codon usage biases tilt the playing field, making certain evolutionary pathways more likely than others?
- And most importantly; if evolution uses such hidden pathways, can we exploit them to design proteins that are resistant to drug escape, or soluble versions of normally intractable membrane proteins?
Our work doesn’t close the story; it opens a new chapter. This paper shows that evolution may have been playing the same game all along: quietly, asymmetrically, and with surprising flexibility. So the question we leave you with is: What else have we missed about how proteins negotiate their evolutionary futures?
Follow the Topic
-
Journal of Molecular Evolution
This journal covers experimental, computational, and theoretical work aimed at deciphering features of molecular evolution and the processes bearing on these features.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in