Taming the uranium(II–VI) ions by a tris(amido)arene ligand
Published in Chemistry

Uranium, the heaviest element abundant in nature, can exhibit multiple oxidation states and rich redox chemistry1. Four oxidation states, uranium(III) to uranium(VI), have been well established. In 2013, Evans and co-workers reported the first molecular uranium(II) complex2. Since then, the low-valent uranium chemistry has seen rapid development in the last decade3, including the recent isolation of a uranium(I) complex4. It is highly desired to stabilise as many oxidation states of uranium as possible in a retained ligand framework, which will allow direct comparison of different oxidation states and controllable redox transformations. Well-defined uranium complexes stabilised by chelating ligands have generated some most exciting chemistry in recent years, including the isolation of a linear, O-coordinated η1-CO2 bound to uranium5 and the first terminal uranium nitride complex6, as well as the electrocatalytic water reduction to produce dihydrogen7. However, no chelating ligand has been shown to support all five well-established oxidation states of uranium, uranium(II–VI). In this study, we aim to design a chelating ligand capable of stabilising uranium(II–VI) with a preserved coordination environment.
Since established in 2017, our research group has focused on studying “f-block metal–arene interactions” and utilizing this concept in exploring new properties and reactivity of f-block metals. We reported the first inverse-sandwich thorium arene complexes8 (Fig. 1a), and unveiled the distinct electronic structures of samarium and ytterbium biphenyl complexes9 (Fig. 1b). Inspired by uranium–arene δ interactions in inverse-sandwich uranium complexes10,11 and Meyer’s tris(aryloxide)arene ligand12,13 as well as reports on π interactions between electrophilic uranium ions and neutral arenes14,15, we anticipated that the ambiphilic nature of arenes might be utilized to balance the stability of low and high-valent uranium ions. In 2021, our group reported two tripodal tris(amido)arene ligands featuring an anchoring arene (as the centre of the 1,3,5-triphenylbenezene backbone) and N-aryl substituents, and introduced them to coordination chemistry of rare-earth metals (Fig. 1c)16. In the current study (https://www.nature.com/articles/s41467-023-40403-w)17, the N-adamantyl version of the tris(amido)arene pro-ligand, H3[AdTPBN3], was prepared and applied to the coordination chemistry of uranium (Fig. 1d). The pre-organized (C3-syn) structure and better crystallinity of [AdTPBN3]3– (compared to the N-aryl analogues) provide ease for work-up and crystallization of uranium complexes. Ultimately, five oxidation states of uranium, i.e., uranium(II–VI), could be stabilised by [AdTPBN3]3– through ambiphilic uranium–arene interactions.
.")
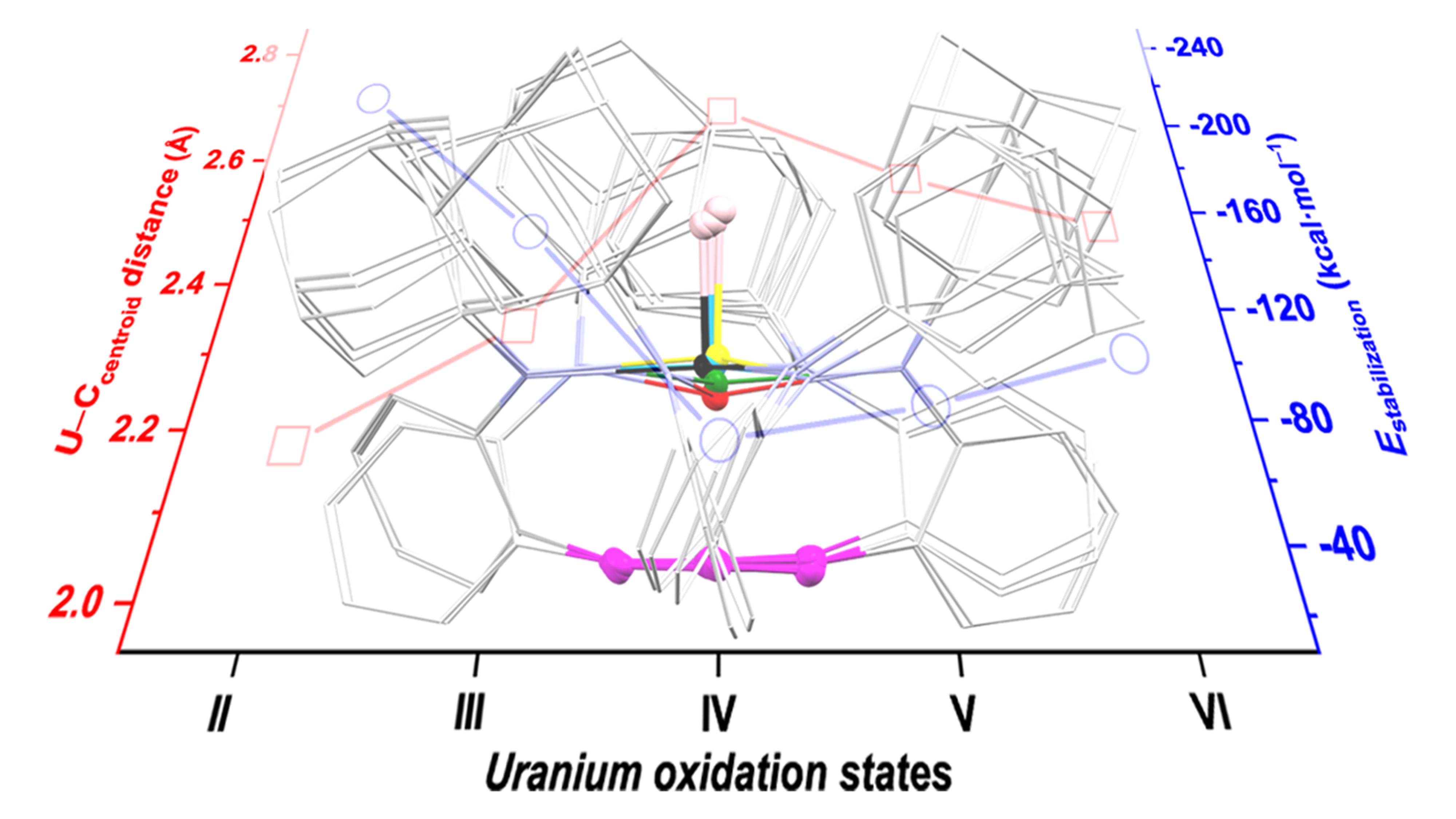
As illustrated in Fig. 2a, deprotonation of H3[AdTPBN3] and subsequent salt metathesis with the uranium triiodide precursor generated the uranium(III) complex 1. While reduction of 1 by KC8 produced the uranium(II) complex 2, 1 could also reacted with oxygen-atom-transfer reagents, such as N2O and pyridine-N-oxide, to furnish the uranium(V) terminal oxo complex 3. 3 could be further oxidized by AgSbF6 or reduced by KC8 to afford the corresponding uranium(VI) and uranium(IV) terminal oxides, 5 and 4, respectively. These compounds were obtained in good yields and exhibited excellent thermal stability under inert atmosphere. For instance, no decomposition of 2 in THF was detected even after prolong heating at 50 ℃. Compounds 1–5 were all characterized by single crystal X-ray diffraction. Notably, 3–5 represent the first trio of structurally authenticated uranium(IV–VI) terminal oxo complexes with the same ancillary ligand. The superposition of molecular structures of 1–5 revealed that the U–Ccentroid distances vary significantly, which peak in the uranium(IV) oxo complex 4 and decrease upon reduction or oxidation (Fig. 2b).
 and molecular structures superposition (b) of uranium(II–VI) complexes 1–5.")
Electrochemical studies were carried out for the uranium(III) complex 1 and the uranium(V) oxo complex 3 to probe their redox properties. Two reversible one-electron redox events were revealed by cyclic voltammetry for each complex (Fig. 3a), matching well with the synthetic findings and suggesting that the direct one-electron oxidation of 1 might be chemically feasible. Reactivity studies (Fig. 3b) indeed showed that oxidizing 1 by AgF or 1,2-C2H4I2 (a convenient substitute of I2) generated uranium halides 6 or 7, respectively. Further oxidation of the uranium(IV) iodide 7 with AgNO2 resulted in the formation of 3. In addition, 1 was converted to the terminal uranium(IV) oxide 4 by reacting with KNO2 in the presence of crypt, while 4 could also be synthesized from the reaction between the uranium(II) complex 2 and oxygen-atom-transfer reagents.
 and redox conversions of 1–5 (b).")
The oxidation state assignment of uranium in these complexes was supported by NMR spectroscopy, electronic absorption spectroscopy, X-ray photoelectron spectroscopy, SQUID magnetometry, and electronic paramagnetic spectroscopy. The experimental characterisation provided the basis for computational studies on their electronic structures. DFT calculations supported the 5f4 (f2δ2) electronic ground state for the uranium(II) complex 2, and also revealed the U–O multiple bonding character of the trio of terminal uranium(IV–VI) oxo complexes 3–5 as well as the potential inverse trans influence therein. To further elucidate the uranium–arene interactions, extended transition state–natural orbitals for chemical valence (ETS–NOCV) calculations were performed on compounds 1–5 with appropriate molecule fragmentation. The σ, π, and δ-type uranium–arene interactions were classified based on the symmetry of the NOCV pairs. The results show that δ backdonation from 5f orbitals (uranium) to π* orbitals (the anchoring arene) dominate in 1 and 2, whereas π donations from π orbitals of the anchoring arene to uranium-based orbitals monotonically strengthen as the oxidation states of uranium increase (Fig. 4a). Notably, the trend of total stabilisation energies of uranium–arene interactions correlate well with the trend of U–Ccentroid distances for 1–5 (Fig. 4b), further supporting our anticipation that the ambiphilic uranium–arene interactions play a key role in stabilising both low and high-valent uranium ions.

In conclusion, a series of uranium(II–VI) complexes supported by a tripodal tris(amido)arene ligand were synthesized and characterised. Controlled one- or two-electron redox transformations could be readily achieved with these uranium complexes. Combined experimental and computational studies supported that the ambiphilic uranium–arene interactions play a pivotal role in balancing the stability of low- (II) and high-valent (VI) uranium ions. We expect that this chelating ligand framework will be a promising platform to explore redox chemistry of uranium in small molecule activations. Such efforts are currently ongoing in our lab. Moreover, the ligand design strategy to incorporate metal–arene interaction may be extended to other metals to stabilise unusual oxidation states and realize challenging redox chemistry.
The funding sources, institutions, and experimental collaborators and supporters have been acknowledged in the original article17. Besides, we also want to thank High-performance Computing Platform of Peking University, where we performed all calculations for this work.
References
1 Löffler, S. T. & Meyer, K. in Comprehensive Coordination Chemistry III Series 3.13 - Actinides (eds Edwin C. Constable, Gerard Parkin, & Lawrence Que Jr) 471-521 (Elsevier, 2021).
2 MacDonald, M. R. et al. Identification of the +2 Oxidation State for Uranium in a Crystalline Molecular Complex, [K(2.2.2-Cryptand)][(C5H4SiMe3)3U]. J. Am. Chem. Soc. 135, 13310-13313 (2013).
3 Boreen, M. A. & Arnold, J. The synthesis and versatile reducing power of low-valent uranium complexes. Dalton Trans. 49, 15124-15138 (2020).
4 Barluzzi, L., Giblin, S. R., Mansikkamäki, A. & Layfield, R. A. Identification of Oxidation State +1 in a Molecular Uranium Complex. J. Am. Chem. Soc. 144, 18229-18233 (2022).
5 Castro-Rodriguez, I., Nakai, H., Zakharov, L. N., Rheingold, A. L. & Meyer, K. A Linear, O-Coordinated η1-CO2 Bound to Uranium. Science 305, 1757-1759 (2004).
6 King, D. M. et al. Synthesis and Structure of a Terminal Uranium Nitride Complex. Science 337, 717-720 (2012).
7 Halter, D. P., Heinemann, F. W., Bachmann, J. & Meyer, K. Uranium-mediated electrocatalytic dihydrogen production from water. Nature 530, 317-321 (2016).
8 Yu, C. et al. Arene-Bridged Dithorium Complexes: Inverse Sandwiches Supported by a δ Bonding Interaction. J. Am. Chem. Soc. 142, 21292-21297 (2020).
9 Xiao, Y. et al. Distinct electronic structures and bonding interactions in inverse-sandwich samarium and ytterbium biphenyl complexes. Chem. Sci. 12, 227-238 (2021).
10 Diaconescu, P. L., Arnold, P. L., Baker, T. A., Mindiola, D. J. & Cummins, C. C. Arene-Bridged Diuranium Complexes: Inverted Sandwiches Supported by δ Backbonding. J. Am. Chem. Soc. 122, 6108-6109 (2000).
11 Liddle, S. T. Inverted sandwich arene complexes of uranium. Coord. Chem. Rev. 293-294, 211-227 (2015).
12 La Pierre, H. S., Scheurer, A., Heinemann, F. W., Hieringer, W. & Meyer, K. Synthesis and Characterization of a Uranium(II) Monoarene Complex Supported by δ Backbonding. Angew. Chem. Int. Ed. 53, 7158-7162 (2014).
13 Halter, D. P., Heinemann, F. W., Maron, L. & Meyer, K. The role of uranium–arene bonding in H2O reduction catalysis. Nat. Chem. 10, 259-267 (2018).
14 Cotton, F. A. & Schwotzer, W. Preparation and structure of [U2(C6Me6)2Cl7]+, the first uranium(IV) complex with a neutral arene in η6-coordination. Organometallics 4, 942-943 (1985).
15 Andreychuk, N. R. et al. Uranium(IV) alkyl cations: synthesis, structures, comparison with thorium(IV) analogues, and the influence of arene-coordination on thermal stability and ethylene polymerization activity. Chem. Sci. 13, 13748-13763 (2022).
16 Xin, T., Wang, X., Yang, K., Liang, J. & Huang, W. Rare Earth Metal Complexes Supported by a Tripodal Tris(amido) Ligand System Featuring an Arene Anchor. Inorg. Chem. 60, 15321-15329 (2021).
17 Deng, C. et al. Accessing five oxidation states of uranium in a retained ligand framework. Nat. Commun. 14, 4657 (2023).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in