The connective tissue protein 'ADAMTSL3' is crucial for heart function
Published in Healthcare & Nursing
Heart failure is a major health concern worldwide. Affecting 2-3% of the population, and 10% of the elderly, the disease drives morbidity, hospitalisation and mortality in patients, with pandemic proportions. As a clinical syndrome, rather than a single diagnosis, the causes of heart failure are multifactorial, and the disease affects a very heterogenous patient population. Although management and treatment of heart failure has improved over the years, full recovery is rare, and the only life-saving cure is cardiac transplantation. This represents a shortcoming in modern medicine, warranting deeper investigation of the underlying pathophysiological mechanisms. In this newly published study, researchers investigate the significance of a protein that resides in-between cardiac muscle cells, ADAMTSL3, in heart failure development, and show how a small component of the cardiac connective tissue can have a great impact on the function of the heart. The study shows how ADAMTSL3 regulates a fine-tuned and crucial signalling response of the heart to pathological stimuli, with potential consequences for future heart failure therapy.
Heart failure arises, in essence, from the impaired ability of the heart to sufficiently pump (systolic failure) or fill (diastolic failure) with blood to meet the body’s need for oxygen. The reasons as to why the heart fails are usually complex, but a number of different risk factors have been identified; major ones being age, obesity, high blood pressure, diabetes, atherosclerosis and cardiac valve disease. Genetic variants in proteins that constitute the contractile apparatus of the cardiac cells are further known to cause heart failure, as heritable cardiomyopathies, or in conjunction with environmental risk factors. Additionally, modulating variants in proteins of other parts of the heart, such as the connective tissue that surrounds the cardiac muscle cells, known as the 'extracellular matrix', are suggested to play a role in heart failure development.
During heart failure progression, the myocardium undergoes extensive remodelling to counteract the harmful stimuli. Changes in pressure or volume load on the heart rallies a range of molecular pathways, with hallmark pathophysiological responses including growth of the cardiac muscle, death of cardiac muscle cells, connective tissue expansion, i.e., fibrosis, and inflammation. In this process, hypertrophic growth often precedes dilatation of the cardiac walls and heart failure. The fibrillar protein matrix in-between the cardiac muscle cells is crucial for maintaining cardiac structural and functional integrity, and an integral part of this remodelling response. Cardiac fibroblasts (CFBs) are the main matrix-producing cells of the heart. During heart failure, CFBs are activated and transdifferentiate into highly proliferating and matrix-producing 'myofibroblasts'. This process is critical in wound healing, however, excessive myofibroblast differentiation drives pathological cardiac remodelling through activation of growth factors, inflammation, reduced cardiac electrical conductance and increased myocardial stiffness, resulting from increased collagen deposition and crosslinking in what is termed cardiac fibrosis. One of the main molecular drivers of CFB activation and cardiac fibrosis is the signalling molecule 'transforming growth factor (TGF)β'. Because of its potent pro-fibrotic properties, TGFβ represents an attractive therapeutic target in heart failure. However, direct TGFβ inhibition is limited by off-target effects, and a better understanding of TGFβ-mediated CFB activation in the heart is needed to improve heart failure outcomes.
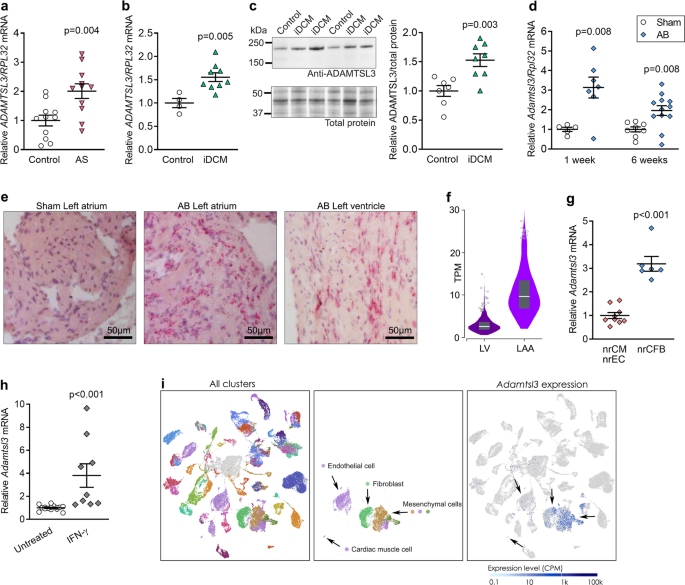
In the study presented here, the role of the matrix glycoprotein ADAMTSL3 (a disintegrin-like and metalloprotease domain with thrombospondin type 1 motifs-like 3') in heart failure was put under investigation. ADAMTSL3 levels were previously shown to be increased in the failing heart, and although its function is largely unknown, the ADAMTSL protein family is suggested to play a role in the regulation of TGFβ. Here, the generation and characterisation of a mouse with targeted inactivation of Adamtsl3 (Adamtsl3 knock-out) is reported, which was used to investigate ADAMTSL3 in the pressure-overloaded heart. One of the most important discoveries were that the knock-outs develop contractile dysfunction and cardiac dilatation, with high mortality, compared to WT mice. This suggests that ADAMTSL3, as a component of the extracellular matrix, plays a crucial role in maintaining the structural and functional integrity of the heart upon pathological alterations in cardiac pressure, which is commonly observed in heart failure patients.
To investigate the molecular mechanism behind the detrimental phenotype of the Adamtsl3 knock-out mice, the researchers performed RNA-sequencing of the mouse heart transcriptome following cardiac pressure overload by constriction of the aorta. This revealed increased TGFβ activity and presence of myofibroblasts in the knock-out hearts compared to WTs, which was confirmed at the protein level. Furthermore, the knock-out had increased cardiac levels of insoluble collagen, indicating increased collagen crosslinking and stiffer cardiac tissue. To test whether ADAMTSL3 regulated the TGFβ-mediated activation of CFBs, ADAMTSL3 levels were experimentally increased in cultures of human CFBs. Here, ADAMTSL3 inhibited TGFβ activity and myofibroblast differentiation, resulting in reduced extracellular matrix production and reduced collagen synthesis.
The findings of this study collectively identify ADAMTSL3 as an important part of the cardiac extracellular matrix, with an essential role in maintaining cardiac function in the failing heart. As a negative regulator of the pro-fibrotic driver, TGFβ, ADAMTSL3 may hold therapeutic potential for heart failure patients, and future investigations will reveal whether ADAMTSL3 has beneficial effects in the failing heart.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
DNA repair and human disease
Publishing Model: Hybrid
Deadline: Oct 31, 2026
Cell death and inflammatory signalling
Publishing Model: Hybrid
Deadline: Oct 28, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in