Unfortunately you are not authorized to view that page.
Uncovering miR-299a-5p as a Driver of Fibrosis in Diabetic Kidney Disease
Published in Biomedical Research
How the Project Began
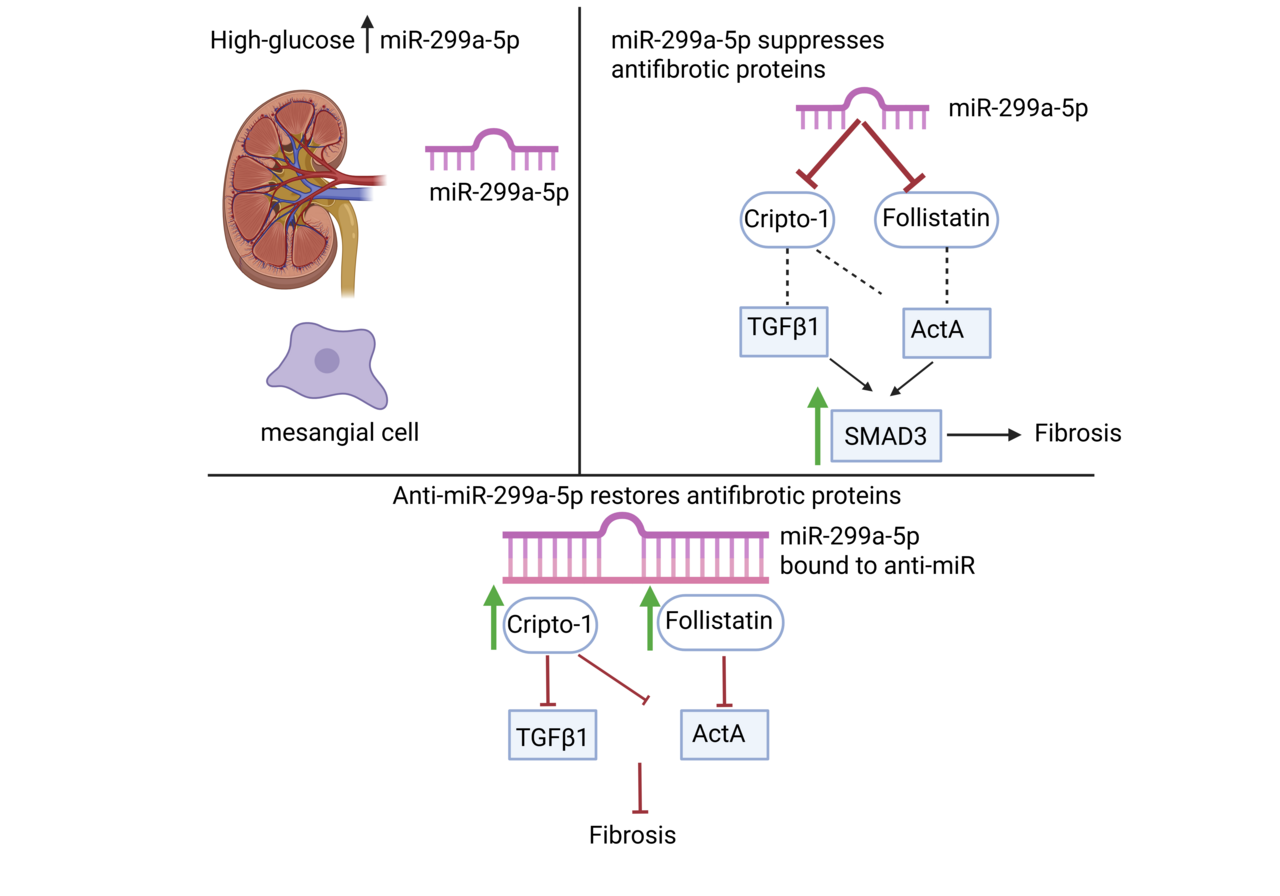
Diabetic kidney disease (DKD) is a common complication of diabetes, developing in up to 40% of patients. Its onset often begins with protein in the urine called albuminuria. Histologically, it is characterized by well-established changes that eventually manifest as fibrosis in all areas of the kidneys. This replacement of the normal kidney structure with fibrosis results in end stage kidney failure that requires dialysis or a kidney transplant to survive1. When this project started, we were motivated by a long-standing question arising in DKD: why fibrosis progresses despite aggressive glucose and blood pressure control, and why established therapeutic targets such as transforming growth factor β1 (TGFβ1) remain so difficult to manipulate clinically2,3. Our previous work revealed that TGFβ1 suppresses the antifibrotic protein follistatin (FST) in mesangial cells by upregulating microRNA (miR)-299a-5p4. That clue became the spark for this study. At the time, almost nothing was known about how miR-299a-5p behaves in the kidney, much less in DKD. But the idea that a single microRNA might simultaneously control multiple antifibrotic pathways was too compelling to ignore.
Early Challenges and the First Breakthrough
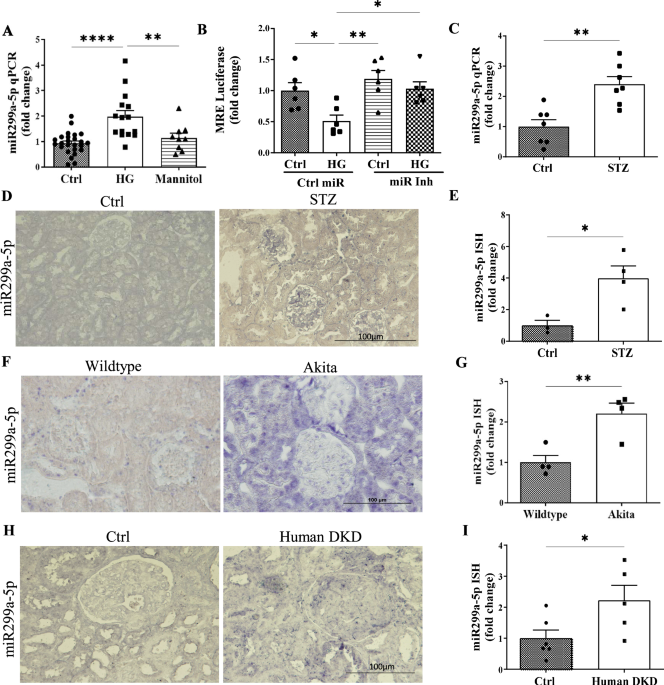
One of the earliest challenges we ran into was simply confirming that miR-299a-5p was relevant in disease. Our first high glucose experiments were surprisingly clear: mesangial cells in the kidney consistently upregulated miR-299a-5p, and mannitol controls reassured us that this was not osmotic stress. The next turning point came when we examined diabetic mouse models. The increase in miR-299a-5p in type 1 diabetic mice was dramatic and immediate, but another model had a more gradual disease course and showed a subtler expression of the miR. This discrepancy initially felt like a setback, because we feared inconsistency across models. Only later did we recognize that this was actually informative: miR-299a-5p expression reflected the tempo of hyperglycemic injury, not a flaw in the biology. That realization shaped how we interpreted the human biopsy data, where chronicity and heterogeneity similarly influenced expression levels5.
But the moment everything changed was when we used the miR-299a-5p response element luciferase reporter. We saw that in the presence of high glucose treatment, there was a drastic reduction in the activity of the response element, indicating an increase in miR-299a-5p expression. Furthermore, we saw a beautiful rescue of luciferase activity when we inhibited the miR. It was the first concrete evidence that we were successfully blocking miR-299a-5p function.
The Unexpected Discovery of Cripto-1
Our lab originally believed FST was the primary antifibrotic target of miR-299a-5p4. Then our bioinformatics screen identified cripto-1 as another predicted target. At first, I honestly doubted it. Cripto-1 was known as a developmental protein and a regulator in cancer, not something we typically associate with the diabetic kidney6–8. But the conservation of the miR-299a-5p recognition site across species was too strong to ignore.
The cripto-1 3’ untranslated region luciferase reporter experiment was one of the most satisfying moments of the entire project. When high glucose reduced cripto-1 reporter activity and the miR-299a-5p inhibitor reversed this, the lab collectively paused, we realized the story was much bigger than we first imagined, as we had reached a “EUREKA” moment.
Equally surprising was discovering that cripto-1 potently inhibited both activin A and TGFβ1 signaling. When we saw Smad3 phosphorylation suppressed by recombinant cripto-1, it became clear that miR-299a-5p acts as a central gatekeeper for multiple fibrotic pathways.
Connecting Caveolin-1, MicroRNAs, and Antifibrotic Signaling
Another behind-the-scenes twist came from our longstanding work on caveolin-1. Years ago, we had noticed that caveolin-1 knockout mice were protected from DKD and showed higher renal FST9,10. Only in this study did we realize that miR-299a-5p was the missing link; caveolae regulate miR-299a-5p levels, which in turn modifies both FST and cripto-1.
Seeing cripto-1 rise in caveolin-1 knockout mesangial cells felt like finding the final piece of a puzzle that had been on our bench for years.
Running the In Vivo Study: Successes, Surprises, and Stress
The 12-week LNA anti-miR treatment in diabetic Akita mice was the most demanding part of the project. We held our breath every time we measured blood pressure and GFR; off-target hemodynamic effects were a real worry, and the mice were hyperactive. Interestingly, the inhibitor had no effect on blood glucose levels, but it did dramatically reduce albuminuria, glomerular hypertrophy, and fibrosis, clinically relevant markers of kidney injury.
One particularly memorable moment was the day we analyzed nephrin expression. Watching podocyte injury markers normalize with miR-299a-5p inhibition was genuinely exciting for the team; it was the first strong indication that we weren’t just affecting matrix accumulation but actually preserving kidney architecture.
Another unexpected finding was the marked reduction of urinary activin A and TGFβ1. These decreases were visually striking in the IHC and reinforced our hypothesis that restoring endogenous antagonists (FST and cripto-1) is more physiologically balanced than targeting TGFβ ligands directly.
What This Work Means for the Future
Throughout the project, we kept coming back to a central theme: microRNAs rarely have a single target, and that complexity can be an opportunity. In this case, miR-299a-5p simultaneously suppresses two antifibrotic proteins, both of which independently regulate key pathogenic cytokines implicated in DKD4,7,8,11–13.
Looking forward, this work opens at least three promising avenues:
-
Therapeutic development
Our data support miR-299a-5p inhibition as a viable strategy to amplify multiple antifibrotic pathways without directly suppressing TGFβ1, which has historically caused adverse effects2. -
Biomarker potential
The rise in circulating miR-299a-5p in diabetic mice aligns with growing interest in serum miRNAs as predictors of DKD severity14,15. -
Tubular biology
Though this study focused on mesangial cells, miR-299a-5p is clearly expressed in tubules. Understanding its role in tubular injury and interstitial fibrosis will be essential.
Final Reflections
This project taught us how a single microRNA can reshape an entire signaling landscape in DKD. The most rewarding part was realizing that by targeting miR-299a-5p, we can restore endogenous renal defenses that the diabetic environment suppresses. It is a shift from blocking disease drivers to restoring kidney homeostasis by empowering the kidney’s intrinsic antifibrotic machinery.
References:
- Sun H, Saeedi P, Karuranga S, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183. doi:10.1016/J.DIABRES.2021.109119
- Voelker J, Berg PH, Sheetz M, et al. Anti-TGF-b1 antibody therapy in patients with diabetic nephropathy. J Am Soc Nephrol. 2017;28(3):953-962. doi:10.1681/ASN.2015111230/-/DCSUPPLEMENTAL
- Gewin LS. TGF-β and Diabetic Nephropathy: Lessons Learned Over the Past 20 Years. Am J Med Sci. 2020;359(2):70-72. doi:10.1016/J.AMJMS.2019.11.010
- Mehta N, Li R, Zhang D, et al. miR299a-5p promotes renal fibrosis by suppressing the antifibrotic actions of follistatin. Sci Rep. 2021;11(1). doi:10.1038/S41598-020-80199-Z
- Jiang L, Li L, Zhang K, et al. A systematic review and meta-analysis of microRNAs in the diagnosis of early diabetic kidney disease. Front Endocrinol (Lausanne). 2025;16:1432652. doi:10.3389/FENDO.2025.1432652
- Strizzi L, Bianco C, Normanno N, Salomon D. Cripto-1: a multifunctional modulator during embryogenesis and oncogenesis. Oncogene. 2005;24(37):5731-5741. doi:10.1038/SJ.ONC.1208918
- Gray PC, Harrison CA, Vale W. Cripto forms a complex with activin and type II activin receptors and can block activin signaling. Proc Natl Acad Sci U S A. 2003;100(9):5193. doi:10.1073/PNAS.0531290100
- Gray PC, Vale W. Cripto/GRP78 modulation of the TGF-β pathway in development and oncogenesis. FEBS Lett. 2012;586(14):1836. doi:10.1016/J.FEBSLET.2012.01.051
- Zhang D, Gava AL, Van Krieken R, et al. The caveolin-1 regulated protein follistatin protects against diabetic kidney disease. Kidney Int. 2019;96(5):1134-1149. doi:10.1016/J.KINT.2019.05.032
- Mehta N, Zhang D, Li R, et al. Caveolin-1 regulation of Sp1 controls production of the antifibrotic protein follistatin in kidney mesangial cells. Cell Commun Signal. 2019;17(1). doi:10.1186/S12964-019-0351-5
- Bianco C, Strizzi L, Mancino M, et al. Regulation of Cripto-1 Signaling and Biological Activity by Caveolin-1 in Mammary Epithelial Cells. Am J Pathol. 2008;172(2):345. doi:10.2353/AJPATH.2008.070696
- Gray PC, Shani G, Aung K, Kelber J, Vale W. Cripto Binds Transforming Growth Factor β (TGF-β) and Inhibits TGF-β Signaling. Mol Cell Biol. 2006;26(24):9268. doi:10.1128/MCB.01168-06
- Soomro A, Khajehei M, Li R, et al. A therapeutic target for CKD: activin A facilitates TGFβ1 profibrotic signaling. Cell Mol Biol Lett. 2023;28(1):10. doi:10.1186/S11658-023-00424-1
- Tariq Z, Hamoudi RA, Mussa BM, et al. Serum microRNAs: A potential blood-based diagnostic biomarkers for diabetic kidney disease in Emirati patients with type 2 diabetes. Diabetes Vasc Dis Res. 2025;22(6):14791641251393720. doi:10.1177/14791641251393719
- Alimena S, Stephenson BJK, Webber JW, et al. Differences in serum miRNA profiles by race, ethnicity, & socioeconomic status: Implications for developing an equitable ovarian cancer screening test. Cancer Prev Res (Phila). 2024;17(4):177. doi:10.1158/1940-6207.CAPR-23-0156
Follow the Topic
-
Communications Biology

An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.
Related Collections
With Collections, you can get published faster and increase your visibility.
Signalling Pathways of Innate Immunity
Publishing Model: Hybrid
Deadline: Feb 28, 2026
Forces in Cell Biology
Publishing Model: Open Access
Deadline: Apr 30, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in