Unleashing the Power of Electron Beams for Unprecedented Chemical Transformations and Nanostructure Fabrication
Published in Chemistry

Gone are the days when chemical reactions were solely initiated by thermal or photochemical means. Today, we are witnessing a flourishing exploration of alternative initiation sources, such as mechanochemistry and electrochemistry, which enable the creation and investigation of new molecules and reaction pathways previously inaccessible. And now, emerging on the forefront of these established methods, we find the focused electron beam—an energy source worth delving into.
Imagine harnessing the power of accelerated electrons in transmission electron microscopes to initiate chemical reactions through scattering. Thanks to remarkable advancements in resolution improvement, including cutting-edge aberration correction techniques, we can now visualize molecules in real-time and witness the marvels of electron-beam-induced processes. We call this technique SMART-EM—short for single-molecule atomic-resolution real-time electron microscopy.
Our initial forays into this exciting realm focused on fullerene C60—a molecule easily manipulated and identified through transmission electron microscopy . For instance, we studied the [2 + 2] cycloaddition reactions between C60 and C70 molecules encapsulated in carbon nanotubes, as well as the fusion of C60 dimers to fullertubes by Stone-Wales rearrangement as evinced through high-speed imaging. In doing so, we unraveled the secrets of several intermediate structures that were previously only theoretical conjectures.
However, groundbreaking was the study on the bottom-up synthesis of fullerene C60, utilizing a tailor-made truxene derivative deposited on a graphene monolayer. This endeavor led us to the astounding discovery of C–C bond formation through the electron beam-induced cyclodehydrogenation reaction, unraveling the selectivity of cove and fjord-region C–H bonds. Through these remarkable achievements, we have unveiled organic reaction mechanisms that mimic their wet-chemical counterparts, empowering organic chemists to harness the expanding repertoire of electron beam-induced reactions in the design of novel carbon nanostructures.
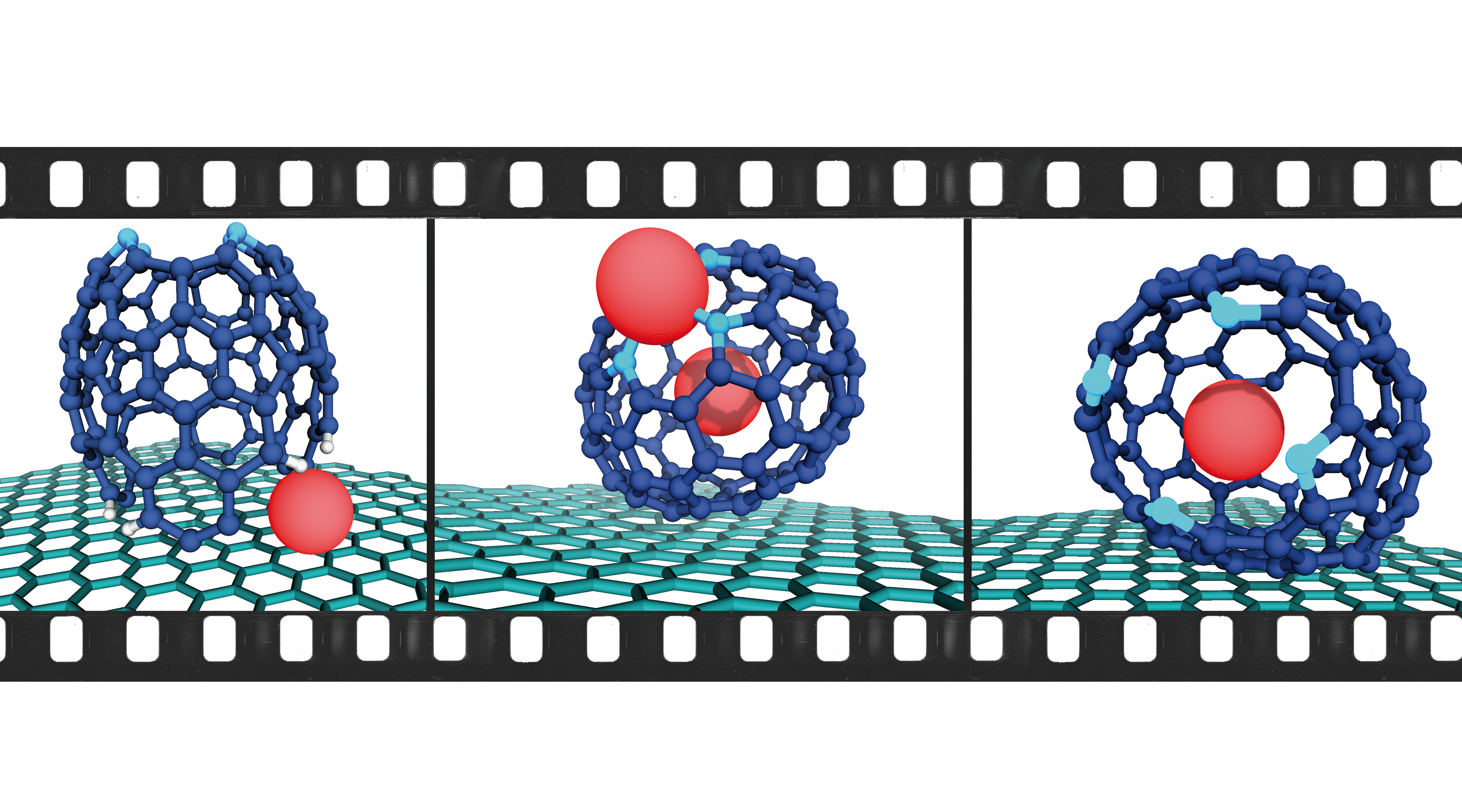
To showcase the remarkable versatility of the electron beam, we ventured beyond established molecular systems and focused on unprecedented structures rooted in the cyclodehydrogenation mechanism we had identified. Our goal was to create an open-cage azafullerene—a captivating fusion of a porphyrin core and a carbon cage architecture.
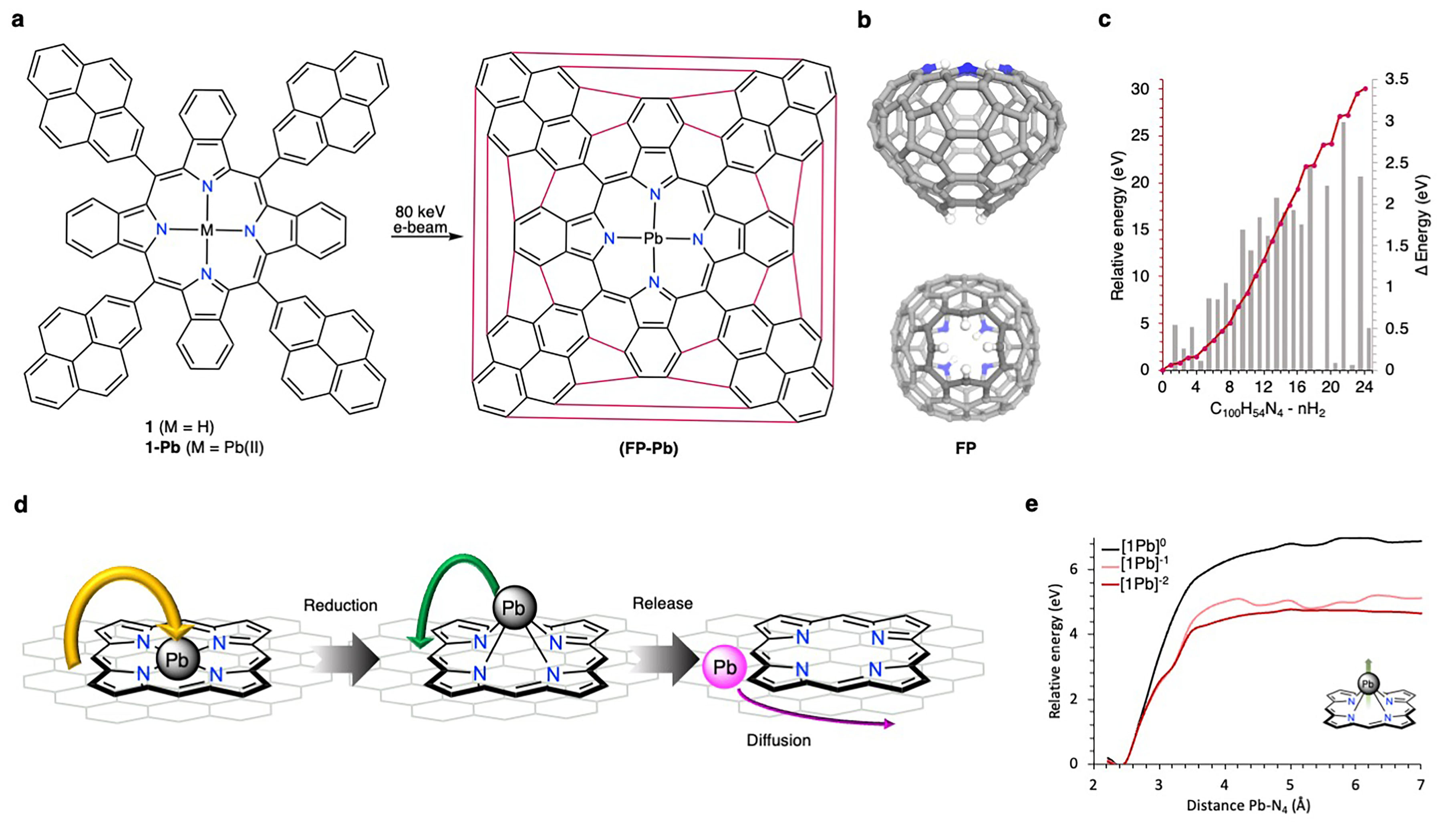
To embark on this endeavor, we synthesized benzoporphyrin 1—a carefully designed molecule adorned with pyrene substituents in the meso-positions, primed to undergo cyclodehydrogenation in a pre-programmed manner, ultimately yielding our desired "fullerophyrin" (FP) (Fig. 1a). To enhance visual contrast, we introduced Pb(II) metalation to the benzoporphyrin precursor, resulting in 1-Pb. The resulting structure of the open-cage FP, with its intricately complexed metal atom, adheres to the isolated pentagon rule (IPR) governing fullerenes, forming a doubly-holed open cage structure (Fig. 1b). Our calculations using density functional theory unveiled a progressive increase in relative energy by approximately 30 eV over the course of 24 cyclodehydrogenation steps (1.25 eV per step), akin to the formation of C60 (Fig. 1c).
To our astonishment, instead of encountering metal-complexed precursors of 1-Pb, we observed loosely diffusing metal atoms on the surface and clusters of Pb atoms forming islands. We rationalized this intriguing phenomenon by postulating a reductive demetallation pathway instigated by the hot-electron reduction resulting from the omnipresent secondary electrons in the periphery of the electron beam, aptly named the beam-shadow (Fig. 1d). Subsequently, the reduced bond strength of the N-Pb bond as 1-Pb transitioned from1-Pb(0) to 1-Pb(-II) lead to the instant demetallation (Fig. 1e).
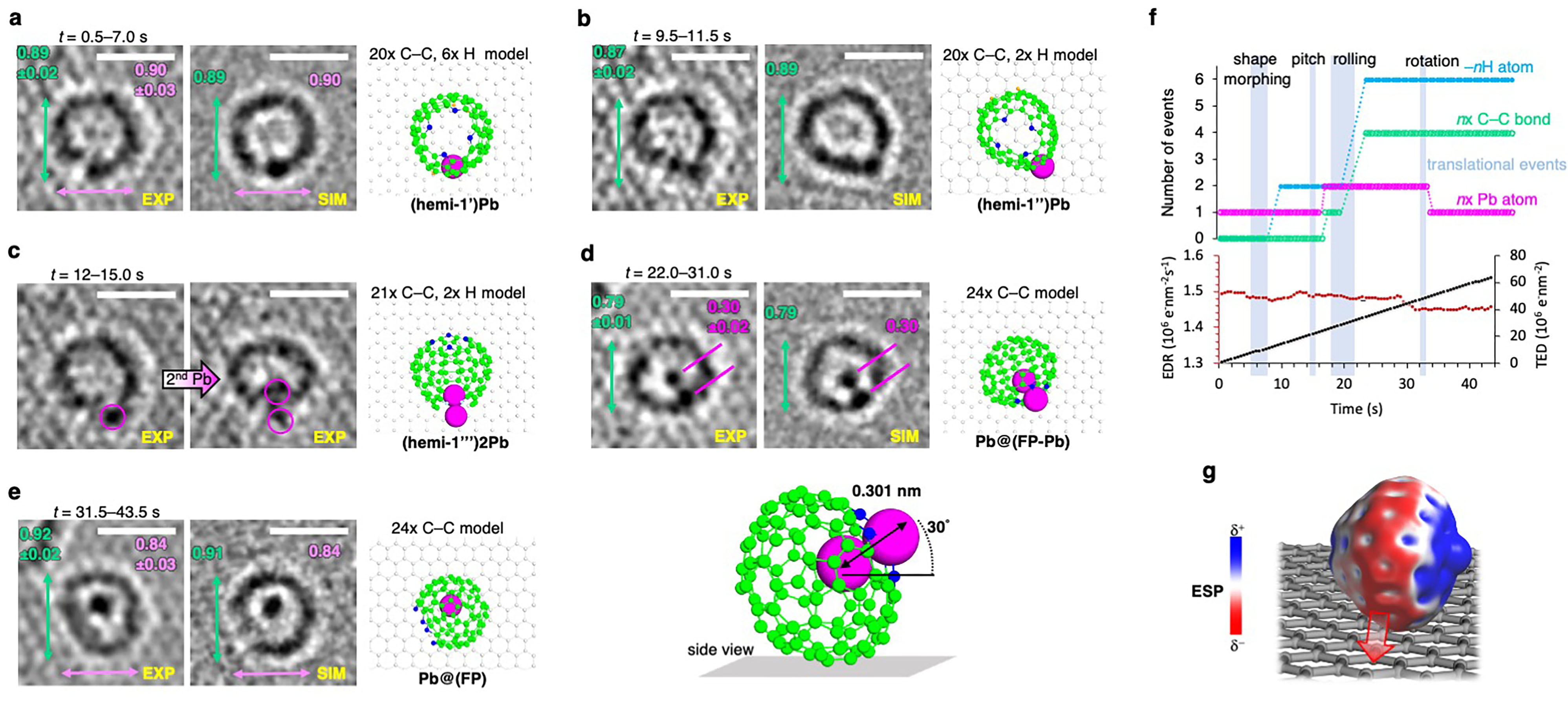
However, amidst our exploration, we encountered a remarkable intermediate structure—(hemi-1’)Pb (Fig. 2a)—that aligned flawlessly with the simulations and calculations in terms of dimensions, shape, and contrast. With the loss of four hydrogen atoms, this structure underwent further transformation, progressing into (hemi-1")Pb (Fig. 2b), which subsequently complexed a second Pb atom (Fig. 2c). After a series of additional transitions on the graphene surface, a fascinating bimetallic cage structure emerged—Pb@(FP-Pb)—with one Pb atom encapsulated within the endohedral cavity and the other complexed by the porphyrinic coordination site (Fig. 2d). Intriguingly, over time, the complexed Pb atom was decomplexed, resulting in the formation of the monometallic species Pb@(FP) (Fig. 2e).
This structure stands out for multiple reasons. Firstly, it demonstrates the remarkable potential for incorporating functional porphyrin cores into carbon cages. Secondly, it represents the first-ever example of both endohedral and skeletal complexation of metal atoms within fullerene-like structures. Lastly, the incorporation of Pb atoms in fullerenes has never been reported before.
The real-time observations have allowed us to establish meaningful correlations between various processes, such as H atom loss or C–C bond formation, and translational events of the structure on the surface (Fig. 2f). Furthermore, we have gained a deeper understanding that beam-molecule interactions can trigger chemically reasonable reaction mechanisms that require less energy, provided the molecular design allows for such mechanisms. Consequently, we only observe physical fragmentation of molecules through knock-on displacements of atoms when energy dissipation via controlled reaction mechanisms is not viable.
Moreover, the chemical binding of the molecule to the surface can be comprehended through the mapping of the electrostatic potential (ESP) of e.g., Pb@(FP-Pb). The high electron density of the outer cage in the metalated species enables chemisorption through [2 + 2] cycloaddition reactions to the surface (Fig. 2g). In contrast, the free base FP, with its lower electron density, frequently dissociates from the surface.
Through deliberate design and a meticulous interplay between theory and experiment, we successfully synthesized and conducted real-time analysis of a hybrid compound using an electron beam. This pioneering endeavor revealed intriguing dynamics of metal complexation. Moreover, alongside confirming the cyclodehydrogenation mechanism, we made a significant discovery regarding the existence of distinct redox regions under the influence of the electron beam. Specifically, an oxidative area was observed in the directly irradiated region, while a reductive area emerged due to the slow secondary electrons at the periphery of the beam.
We firmly believe that what we have uncovered thus far is merely the tip of the iceberg when it comes to the vast array of reaction mechanisms that can be induced by the electron beam. Consequently, exploring radiation chemical events and elucidating the underlying reaction mechanisms will serve as a catalyst for harnessing the focused electron beam's potential in precisely transforming and fabricating novel carbon nanostructures from precisely defined precursor materials.
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in