Unlocking the Catalytic Potential of DNA: Insights from a Structure of the RNA-Cleaving 10-23 DNAzyme
Published in Cell & Molecular Biology


I have always been fascinated by nucleic acids that can perform chemical reactions. As an undergraduate, when I first learned about catalytic RNA molecules called ribozymes, I was immediately drawn to studying them in the lab. This experience changed the course of my life, and when I established my own research group at West Virginia University, I knew I wanted to venture into an even more intriguing area: catalytic DNA molecules known as DNAzymes.
Typically, we think of DNA as inert, serving as a storage unit for our genetic information. However, there are certain types of DNA evolved in the laboratory that defy the conventional rules. These DNAs can fold into complex shapes, enabling them to perform a remarkable range of reactions. For example, they can act as molecular scissors with precise specificity to cut RNA or DNA, or they can function as glue to covalently bind nucleic acids together. DNAzymes possess unique properties that make them highly attractive for various biotechnological applications. They exhibit enzymatic turnover, can specifically target molecules through hybridization, and offer advantages such as inherent storage stability and low synthesis costs. Moreover, DNAzymes hold great promise for applications beyond the research lab, including therapeutic interventions and diagnostic tools. Unfortunately, until recently, the lack of structural information about DNAzymes hindered their full potential from being realized.
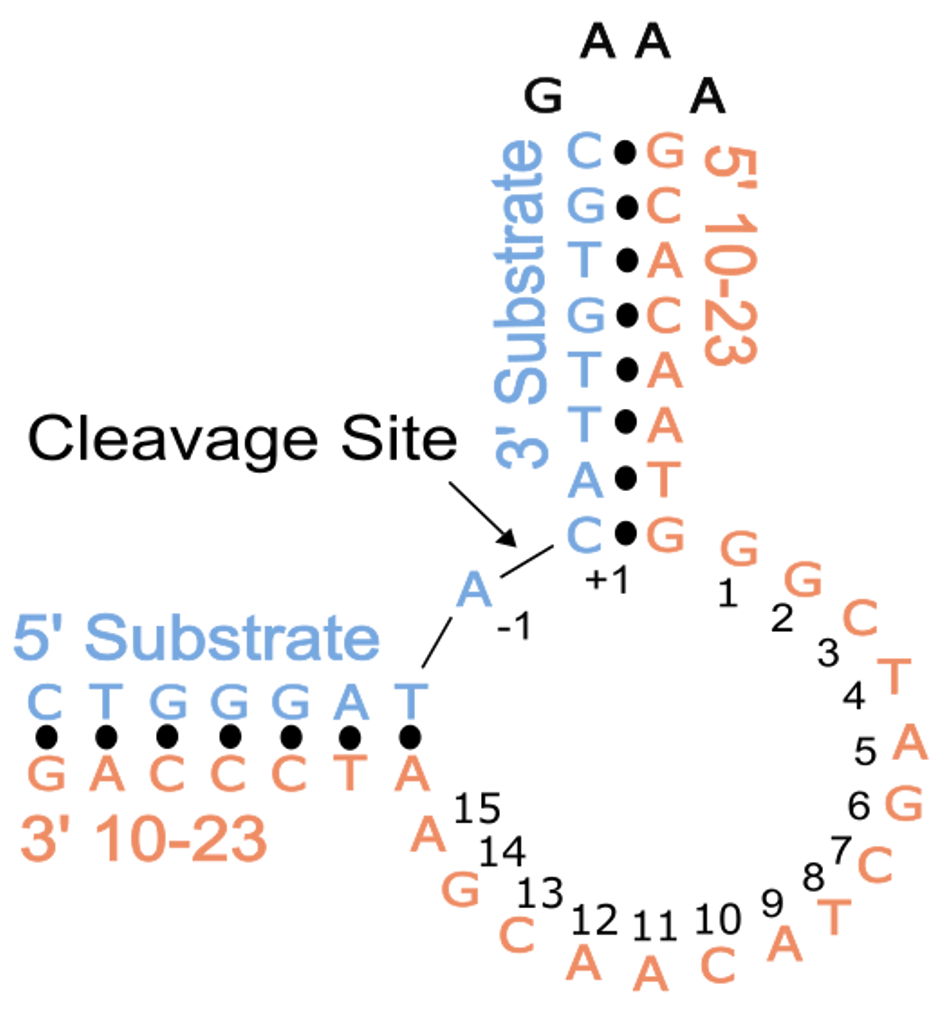
In this study, conducted in collaboration with the Northeastern Collaborative Access Team (NE-CAT) at the Advanced Photon Source (APS), we aimed to overcome the challenges associated with crystallizing DNAzymes, which often result in misleading artifacts. To achieve this, we designed our experiments by placing the substrate and DNAzyme together on a single strand of DNA using a snapback tetraloop. This arrangement allowed the cleavage site in the substrate to align within the catalytic core of the DNAzyme. Indeed, in vitro experiments confirmed that this construct supported the desired activity. To capture the structure of the pre-catalytic 10-23 DNAzyme, we introduced a modification by replacing the 2'-OH position of the RNA nucleophile with a 2'-O-methyl (2'-OMe) group. This modification inhibits the activation of the nucleophile and, consequently, the DNAzyme's catalytic activity.
 Crystallizing nucleic acids is notoriously challenging due to their highly negatively charged backbone and limited chemical diversity compared to protein side chains. To address this, we employed a strategy previously used for the related 8-17 DNAzyme, where we bound the unique blunt DNA end of our crystallization construct to the African Swine Fever Polymerase X (Asfv polX). The protein acted as a chaperone for crystallization and provided a template for solving the crystallography "phase problem" using a method called molecular replacement.
Crystallizing nucleic acids is notoriously challenging due to their highly negatively charged backbone and limited chemical diversity compared to protein side chains. To address this, we employed a strategy previously used for the related 8-17 DNAzyme, where we bound the unique blunt DNA end of our crystallization construct to the African Swine Fever Polymerase X (Asfv polX). The protein acted as a chaperone for crystallization and provided a template for solving the crystallography "phase problem" using a method called molecular replacement.
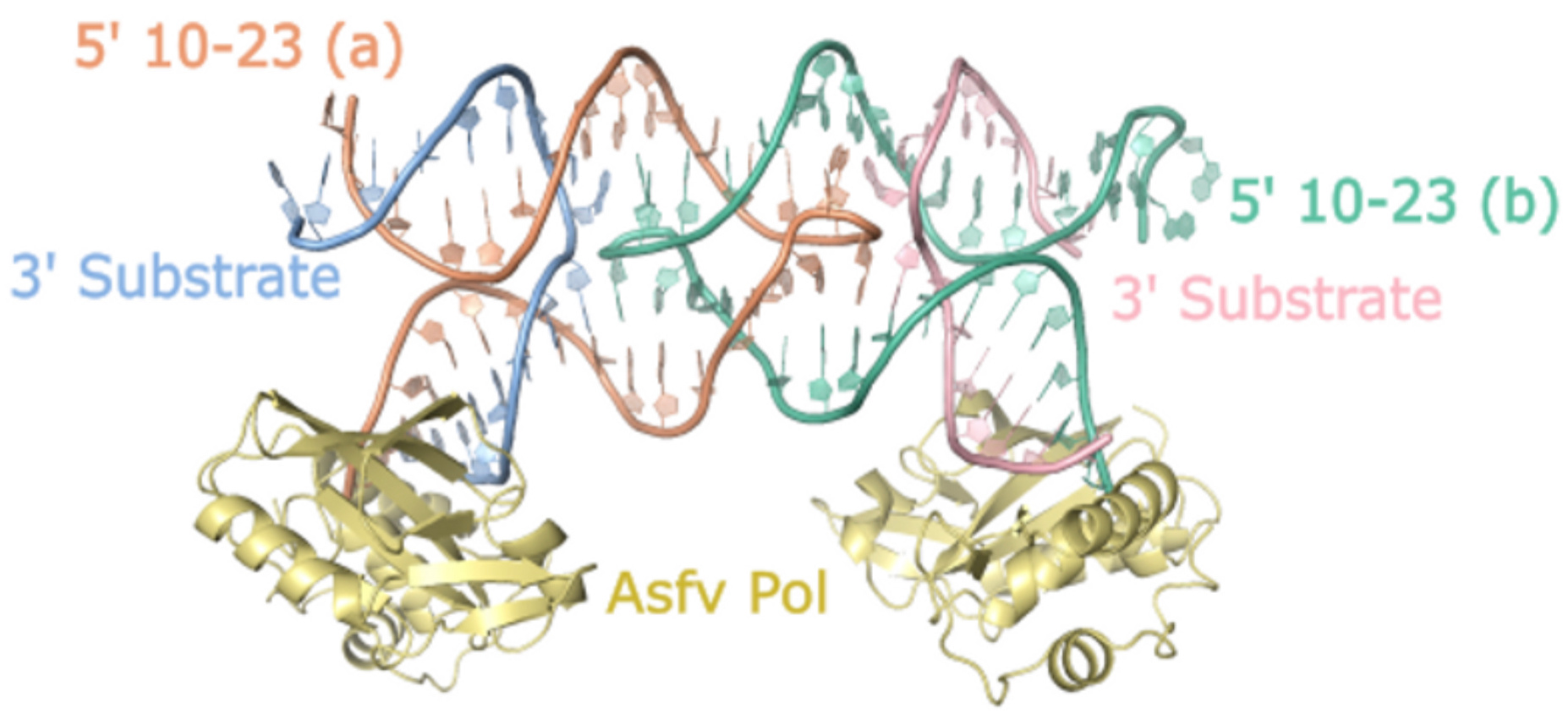
Through our efforts, we successfully determined the crystal structure of the 10-23 DNAzyme, captured in a homodimer conformation. The dimerization we observed was primarily mediated by palindromic sequences within the catalytic core sequence of the DNAzyme. In solution, the dimer was present but represented a minor population, suggesting that the observed dimer conformation likely does not correspond to the true catalytically active form. Nevertheless, this structure provided valuable insights into the coordination of the DNAzyme substrate and the potential involvement of critical catalytic magnesium metal ions.
The structure unveiled a characteristic feature of RNA-cleaving enzymes: the bending of the substrate at the cleavage site. However, the crystal structure did not capture the alignment of the nucleophile with the scissile phosphate, which is essential for catalytic activity. To achieve this alignment, a base flip of an active site purine would be required. Therefore, the stable but improperly organized state of the catalytic core may be a key factor that slows down the overall reaction rate of the 10-23 DNAzyme and renders it more dependent on high concentrations of magnesium cofactor. Consistent with this, we observed three magnesium ions near the scissile phosphate, which has been suggested previously for the 10-23 DNAzyme. Two magnesium ions were found to coordinate with the 2'-OH nucleophile, the 3' bridging oxygen, and the 5' leaving group of the cleavage site. The additional metal ion was coordinated to a non-bridging oxygen on the scissile phosphate. While magnesium is known to be a required cofactor for the 10-23 DNAzyme, the precise mechanism of magnesium's participation in the reaction remains uncertain and necessitates further structural investigations to fully elucidate the RNA cleavage activity.

Where do we go from here? Our journey using X-ray crystallography to study the 10-23 DNAzyme led us to an unexpected dimer, which we had specifically designed our crystallization construct to avoid. Despite our best efforts, the dimeric crystal contact limits the scope and interpretation of the study. Therefore, alternative methods will be crucial to fully understand the dynamics of the 10-23 DNAzyme and to capture a pre-catalytic in-line attack structure. Obtaining pre-, post-, and intermediate structural views of the 10-23 DNAzyme will enable us to understand the interactions necessary for active site organization and target site cleavage. This knowledge could facilitate the rational design of strategies to stabilize the active conformation of the 10-23 DNAzyme and enhance its overall catalytic efficiency, allowing for broad applications as a therapeutic and diagnostic tool for various human diseases.
Follow the Topic
-
Communications Chemistry

An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
f-block chemistry
Publishing Model: Open Access
Deadline: Feb 28, 2026
Experimental and computational methodology in structural biology
Publishing Model: Open Access
Deadline: Apr 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in