Unraveling the Hidden Complexity of PRPS Enzymes
Published in Ecology & Evolution, Cell & Molecular Biology, and Genetics & Genomics
Phosphoribosyl pyrophosphate synthetase (PRPS) enzymes may sound like a mouthful, but these are some of the cell’s most indispensable workhorses. They are responsible for producing PRPP (phosphoribosyl pyrophosphate), a crucial building block for the nucleotides that make up DNA and RNA. Without PRPS, life as we know it simply couldn’t exist. These enzymes are found in nearly every living organism, with origins dating back to the last universal common ancestor (LUCA). Their importance first came to light in the 1950s, when Arthur Kornberg and colleagues identified PRPP and partially purified PRPS.
By the 1970s, deeper biochemical studies revealed that purified PRPS enzymes tend to form aggregates in vitro. This led to the idea that inside cells, PRPS might not act as isolated units but as larger complexes, sometimes weighing more than 30 times the individual protein size. Around this time, a clinical link emerged: patients with excess uric acid due to PRPP overproduction were found to have PRPS superactivity, an X-linked inherited disorder. Studies of PRPS from these patients pointed to structural defects in the enzyme as the cause, pushing PRPS into the biomedical spotlight.
In the late 1980s, researchers in Japan cloned the first three human PRPS genes: PRPS1, PRPS2, and the testes-specific PRPS1L1. Soon after, the same group showed that purified PRPS from rat liver appeared as a complex of four distinct components, two of which were PRPS1 and PRPS2. In the early 1990s, the other two were identified as PRPS-associated proteins 1 and 2 (PRPSAP1 and PRPSAP2). Interestingly, both AP1 and AP2 seemed to lack the residues needed for enzymatic activity. Instead, they appeared to act as regulators, since removing them from enzyme complexes altered PRPS activity in vitro.
The mid-1990s marked a turning point with the discovery of the first point mutations in PRPS1 in human patients, expanding our understanding of how genetic variants in PRPS1 contribute to disease. Since then, dozens more PRPS1 mutations have been identified, widening the spectrum of PRPS1-related disorders. Structural biology helped explain some of the effects of these mutations, with the first bacterial PRPS crystal structure solved in 2000, and the human structure following in 2006. This revealed that PRPS1 assembles as a hexamer with distinct active and allosteric sites. More recently, cryo-electron microscopy (cryo-EM) studies have shown that these hexamers can stack into long filaments, hinting at further regulatory complexity.
A new chapter began in 2014, when my advisor, Dr. Cunningham, showed that oncogenes like Myc can specifically upregulate PRPS2 in lymphoma, highlighting a distinct role for PRPS2 in cancer metabolism. Since then, the field has largely split into two camps: researchers studying PRPS1 as a standalone enzyme in the context of disease mutants, and others investigating PRPS2’s involvement in cancer. However, very few have considered PRPS1 and PRPS2 together, and almost no one has examined the roles of their partners, the PRPSAPs, in this context.
Despite decades of research, several longstanding mysteries remained. Even with structural insights into PRPS1 itself, why do we still lack a clear understanding of how certain PRPS1 mutations impact enzyme activity? Why do mammals retain so many PRPS paralogs, even those that lack catalytic activity? How do these components work together inside cells? What are the precise functions of PRPSAPs? When I joined the lab, these questions drew me in. Were these non-catalytic PRPSAPs (zombie enzymes) evolutionary relics, or do they play a hidden, essential function?
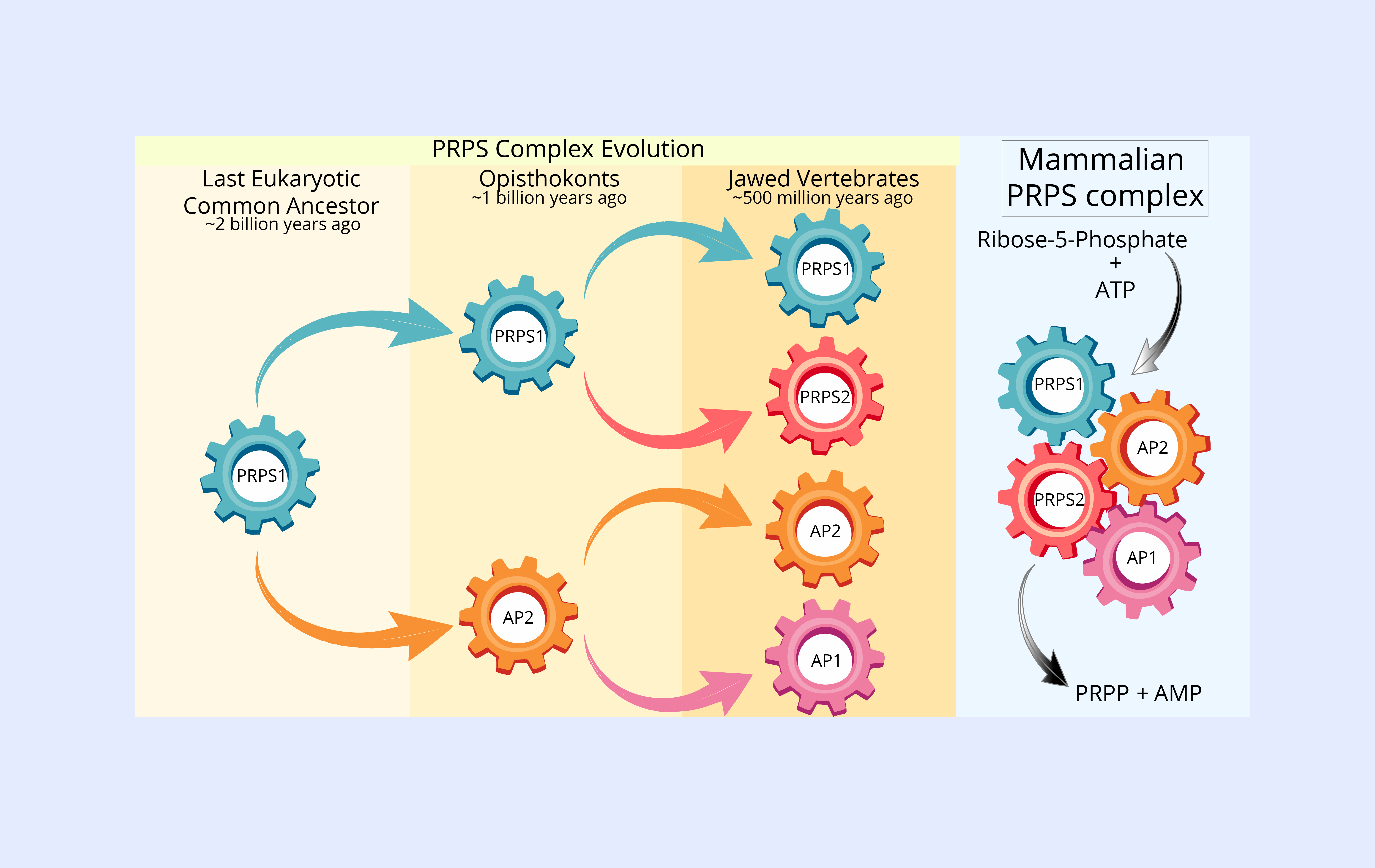
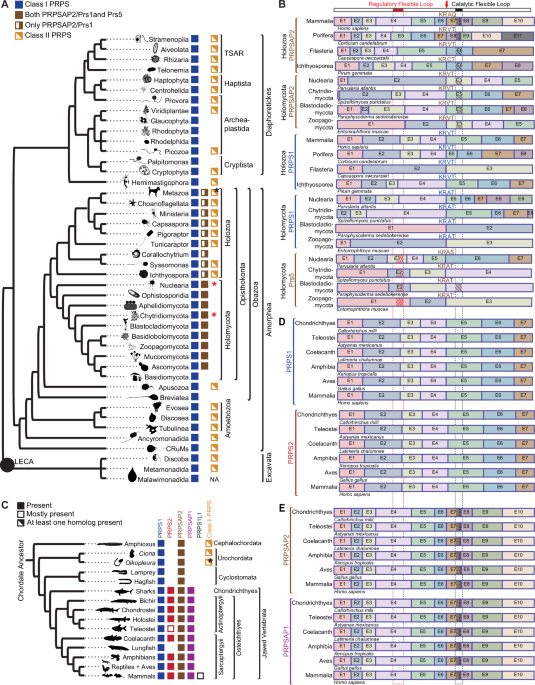
To answer this, we turned to evolutionary genomics. Our analysis revealed that PRPS1 is the original form of the enzyme, introduced into eukaryotes through a bacterial ancestor billions of years ago. The earliest gene duplication produced AP2, which emerged in the common ancestor of animals and fungi over a billion years ago. Much later, a genome duplication event led to the appearance of AP1 and PRPS2 from AP2 and PRPS1, respectively, changes that coincided with the evolution of jawed vertebrates. Each of these paralogs has been retained for hundreds of millions of years, each with unique properties and functions, not just redundant backups. We were also surprised to find that many other eukaryotes, including plants, amoebozoans, and protists, have experienced similar gene duplication events and have retained multiple PRPS paralogs. This recurring evolutionary pattern across diverse lineages provided further evidence that the mammalian PRPS paralogs likely serve important functions, reinforcing the idea that their unique roles are well worth investigating.
To better define the roles of each PRPS paralog, we used CRISPR-Cas9 gene editing to systematically knock out different combinations of these genes in mammalian cells. In every scenario, loss of any PRPS protein led to reduced cellular fitness. Cells expressing only PRPS1 were especially impaired. They grew more slowly, produced fewer nucleotides, and displayed mitochondrial defects, showing that PRPS1 alone is not enough to meet the cell’s metabolic demands.
To uncover the basis of these functional differences, we used a range of biochemical and biophysical methods to map how the PRPS proteins interact and assemble. We looked at which subunits bind to each other, which combinations are favored, the order of assembly, and the resulting configurations when one or more components are missing. Our experiments showed that all four major PRPS paralogs interact to form a large multi-enzyme complex within cells as big as the largest known metabolic assemblies. When AP1, or to a lesser extent AP2, was missing, this complex failed to assemble properly, highlighting their crucial roles as scaffolding proteins that promote higher-order assembly.
We found that the organizational principles of this enzyme complex are more intricate than previously appreciated. The PRPS proteins can assemble into a variety of heterogeneous configurations, not just a single defined structure. We also showed that this complex adopts tissue-specific configurations in mice, suggesting that different cell types might favor particular assembly states to fine-tune PRPS activity in response to metabolic needs. Notably, PRPS1 can form homo-oligomers by itself—something suggested decades ago and confirmed in recent cryo-EM studies—but these PRPS1-only assemblies are not sufficient to support normal cell growth.
Through these experiments, we were able to map the basic blueprint of the PRPS complex for the first time, shedding light on how each component contributes to the complex’s stability and function within the cell. These discoveries have important implications for disease. For example, limiting PRPS activity could potentially restrict nucleotide production to reduce tumor growth, while boosting activity could help patients with nucleotide deficiency syndromes. By understanding the structure and assembly of the PRPS complex, we lay the groundwork for developing new diagnostics and treatments that have the potential to enhance patient health.
Our work underscores that PRPS enzymes are far from simple cogs in the cellular machinery. Instead, they are part of a highly tuned, evolutionarily conserved system for managing life’s most essential building blocks. Understanding how these components assemble and regulate each other opens new possibilities—not just for basic biochemistry, but for understanding metabolic disorders, cancer, and the evolution of complex life.
Being involved in this research has been both humbling and inspiring. One key takeaway for me is that some of the most exciting scientific discoveries happen when we look past what’s assumed and start questioning the familiar. Challenging conventional wisdom can reveal unexpected insights and open new paths for understanding the world.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in