Unveiling the interaction patterns governing binding specificity between proteins
Published in Chemistry, Physics, and Protocols & Methods
Proteins like machines methodically work day and night to maintain physiological functions in the body. The orderly manner of proteins’ activities in the crowded cellular context seems like that there are instruction transmissions in an highly organized society. We are always curious about how proteins at molecular level recognize their partners specifically and transmit information precisely. A more fundamental question is how evolution shapes protein-protein interactions to function in an orderly manner within cellular context. These questions drive us to integrate the studies of protein folding, binding and evolution at molecular level.
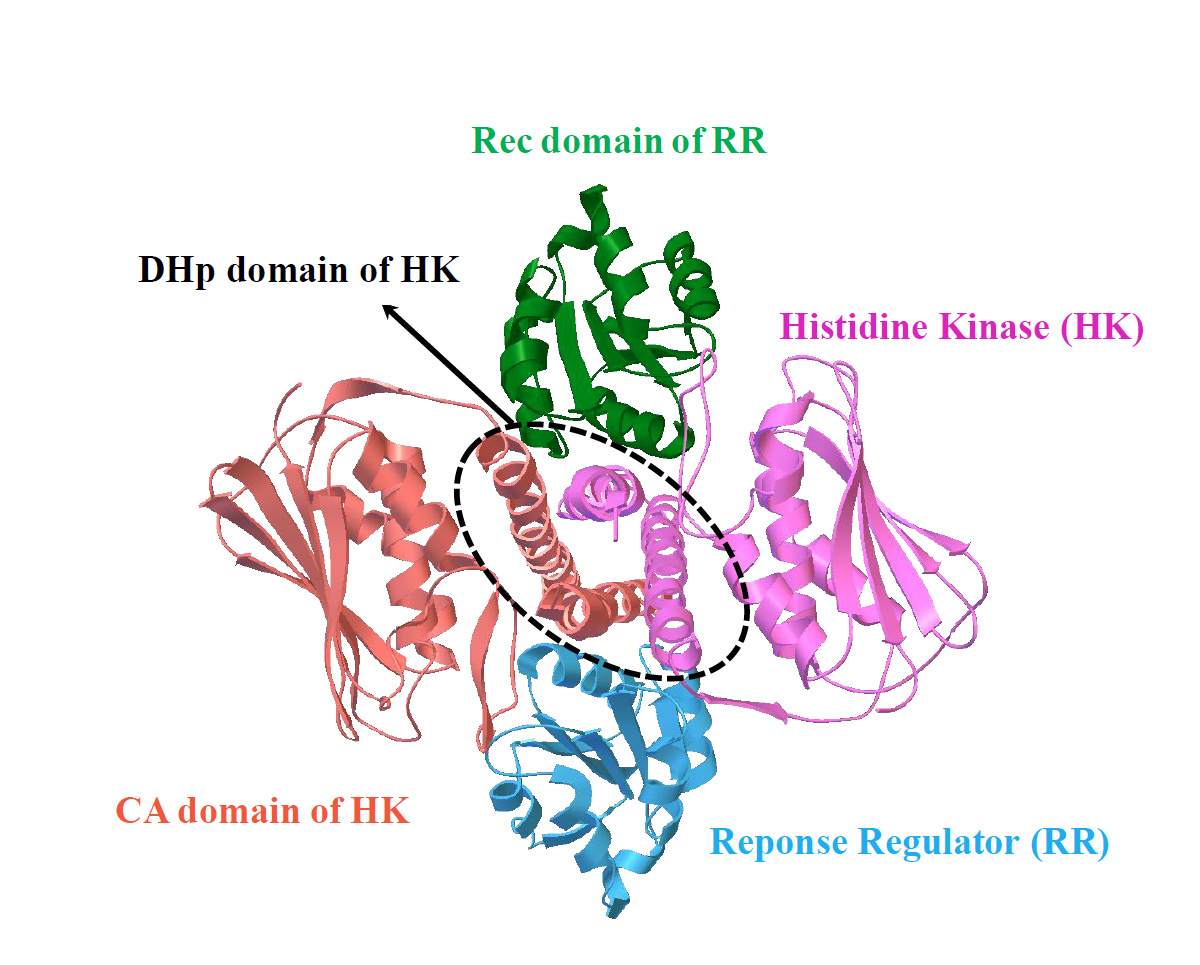
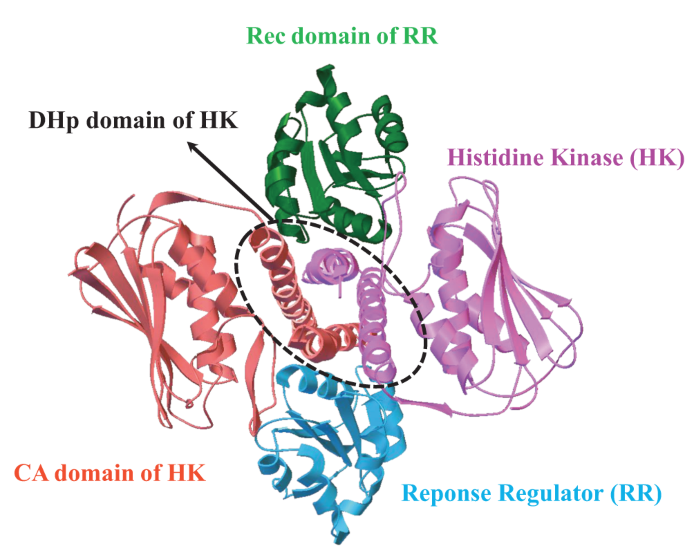
Our work, recently published in Communications Chemistry, aims to unveil the underlying rules of interaction patterns that determine the binding specificity between protein partners. Our hypothesis is that the interaction patterns can be extracted from the evolutionary information imprinted on naturally occurred homologous sequences, as well as computational-simulated evolved sequences. We believe that naturally occurred homologous sequences store the hints of common family-wide interaction patterns for protein stabilities and functions, while structure-oriented computational evolved sequences carry the structure-specific interaction patterns which are tuned to bind cognate partners. To validate our hypothesis, we chose an ideal protein complex model, two-component signaling (TCS) system, which is the most prevalent signal transduction system in bacterial for sensing and responding to environment stimuli (Fig. 1).
")
entry:3DGE). TCS contains a cognate protein pair, i.e. histidine kinase (HK)
and response regulator (RR); the complex structure of TCS is formed by
one HK dimer and two RR monomers; HK dimer is composed of one
histidine phosphotransfer (DHp) domain, and two catalytic and ATPbinding
(CA) domains; RR is composed of receiver (Rec) domain.
In silico, we reconstructed the history of protein evolution in the sequence space by developing a protein evolution simulation method according to the well-established and validated funneled energy landscape theory [1-2]. Similarly as protein folding in the structural space, protein evolution in the sequence space follows a bawl-like energy landscape (Fig. 2). Computational-simulated evolved sequences from the basin of the bawl-like energy landscape are those carrying the structure-specific interaction patterns which are tuned to bind cognate partners.

The basin means the size of the sequence entropy for the evolved sequences.

Having naturally occurred homologous sequences and structure-oriented evolved sequences for analysis, we observed three rules of interaction patterns that could regulate the binding specificity, at least for TCS system. Firstly, highly conserved amino acid positions locate at the binding surfaces for functional needs. This suggests functional requirements are more structure-specific than folding stability. Secondly, a few positions, even physically remote to each other, are highly coupled in variation of amino acids. Surprisingly, the frequency-magnitude of coupling conservations within the structure follows a power law distribution. In addition, those positions with highly coupling conservation constitute an interaction network connecting the binding surfaces for functions and hydrophobic core for folding stability. Thirdly, to realize the binding specificity between cognate partners, additional positions are tuned to adapt to the binding partner. These tuned interaction patterns are complementary to the common family-wide ones.
Back to the questions, our work have dig out the interaction patterns that regulate the binding specificity between proteins and emphasized the evolutionary power in shaping the interaction patterns for protein stability and functional binding. By connecting the protein folding, binding and evolution through the analysis of interaction patterns, our methods and findings shed light on the structure prediction and design of protein complex, which could lead to evolutionary design of novel cognate protein partners. At last, we invite readers to look into our published paper for a more detailed exploration and potential following research of our findings.
Reference
- Z. Yan, J. Wang, Funneled energy Landscape unifies principles of protein binding and evolution. Proc. Natl. Acad. Sci. U.S.A. 117 (44) 27218-27223, (2020).
- Z. Yan, J. Wang, Superfunneled energy landscape of protein evolution unifies the principles of protein evolution, folding and design. Phys. Rev. Lett. 122, 018103 (2019).
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in