When a big heart isn’t a good thing
Published in Chemistry, Cell & Molecular Biology, and General & Internal Medicine
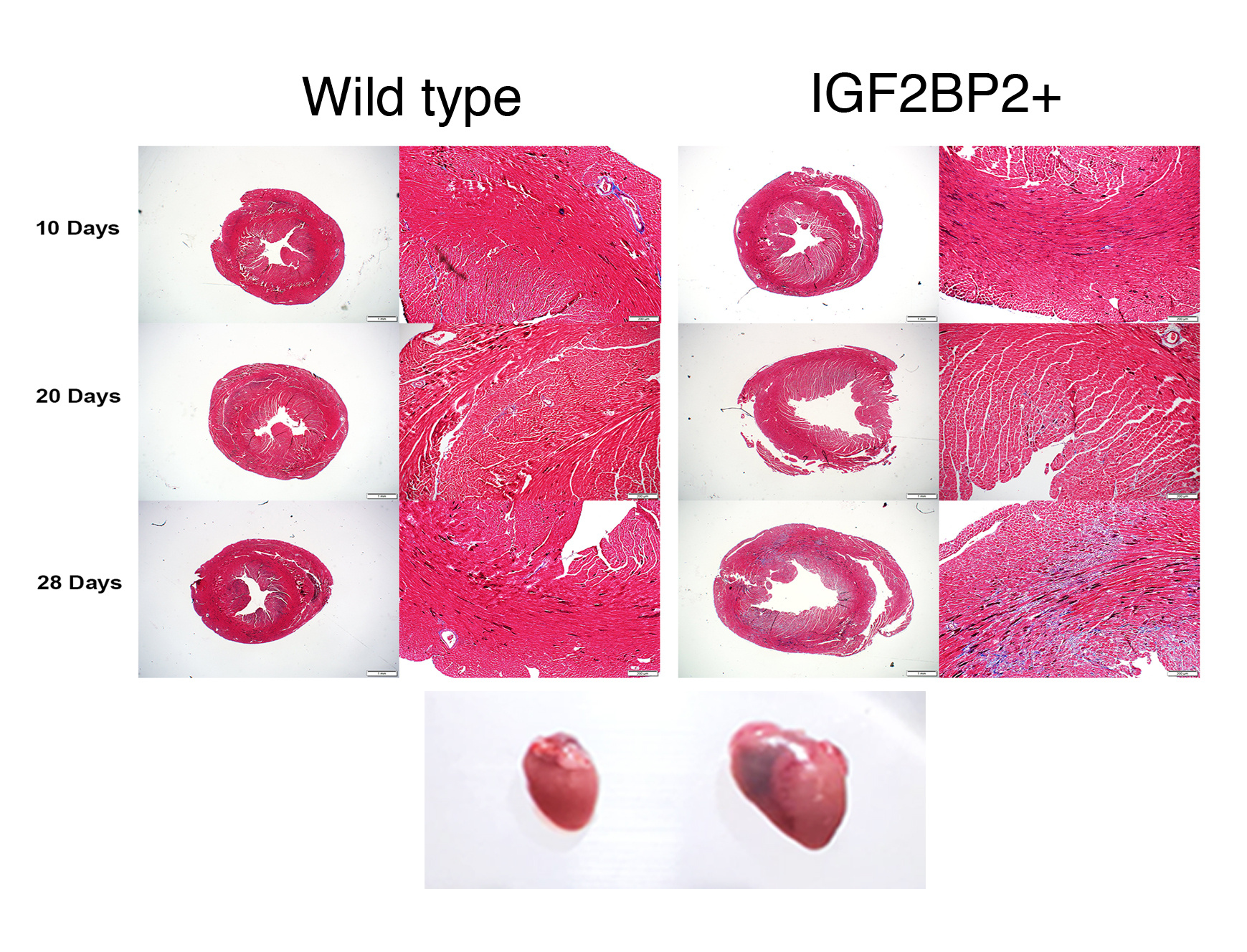
When the student taking care of our mouse lines told me, “You have to see this,” I knew something was unusual. We opened a cage and watched while the mouse circled around, looking normal. All of a sudden, he froze, keeled over, and died. The student told me that the mice of this particular genetic cross had suddenly begun to die in this way over the last day. This mouse had been genetically engineered to express an RNA binding protein on which we work, IGF2BP1, in all the cells of the body. Normally, this protein is expressed in many cells of the embryo, but after birth, the expression drops to almost nothing throughout virtually the entire body. In many kinds of cancers, this protein is again turned on and is associated with tumor progression and metastasis. This was, in fact, one of the reasons we had been studying the protein. These mice, however, were only about 4 weeks old, and seemed much too young to be dying, suddenly, of anything associated with cancer. Knowing nothing of mouse forensics, we consulted with some experts, who suggested that this sounded very much like something connected to the heart. When we performed an autopsy on these mice, we very quickly realized that the heart was twice the size of hearts from litter mates not expressing the gene.
(3D modelling was done by Yuval Cinnamon at the Volcani Institute, using High-Resolution Episcopic Microscopy (HREM). WT, wildtype; VICKZ-TG, IGF2BP1-expressing heart).
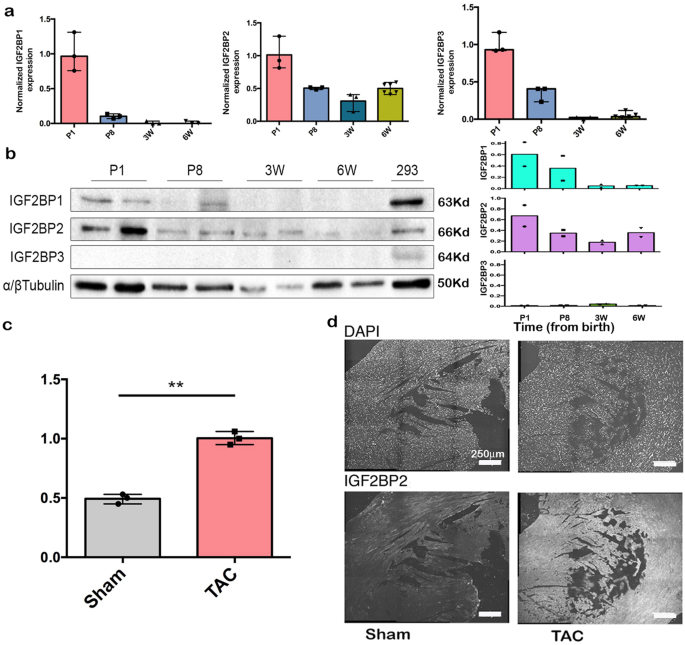
Sudden cardiac death syndrome is a devastating consequence of dilated cardiomyopathy (DCM), a condition in which one or more of the chambers of the heart enlarge in order to compensate for reduced effective pumping. DCM can occur as a result of genetic causes, i.e., mutations in genes essential for the proper organization and function of the heart, or as a result of some sort of stress to the heart, such as might occur when the arteries narrow (pressure overload) or when oxygen supply to cardiac muscles is impaired (ischemia). When we went through the literature looking at genes that were upregulated in DCM, we were initially surprised to discover that it was the IGF2BP2 paralog that was consistently associated with the condition in a number of studies. IGF2BP1, which was known to be an oncofetal protein, was not expressed or turned on by stress in the adult heart. IGF2BP1 and 2 are very similar proteins, however, and share 40% of their RNA targets; thus, they might be expected to function similarly when activated in the same cells. We hypothesized that activating IGF2BP1 in the transgenic mouse line we used was perhaps phenocopying what happens when IGF2BP2 is activated, such as by stress. To test this hypothesis, we collaborated with Sonja Kessler at the Martin-Luther University Halle-Wittenburg, who provided us with a transgenic mouse in which IGF2BP2 expression could be directed to just the heart and turned on or off as desired.
These experiments showed us that IGF2BP2 indeed caused the same DCM phenotype in mice, whether it was turned on before birth or only after the mice became adults. In either case, all of the mice died within 3-4 weeks of activating the gene, all with the same problems of heart function and DCM morphology. Much to our surprise, when the protein was turned on for only 2 weeks and then turned off, all of the mice recovered, and by 12 weeks the hearts were back to normal. The window of opportunity for rescuing the mice was very short, however, and prolonging IGF2BP2 expression for just a few more days already led to a mixed response, with only 2 out 5 mice fully recovering. These results suggested that the DCM morphology induced by IGF2BP2 is potentially reversible, if caught in time.
To study the mechanism behind how IGF2BP2 causes these changes, we performed a mass spectrometry analysis on hearts from wild type and IGF2BP2-expressing mice. A comparison of the proteomes revealed that many mitochondrial and sarcomeric proteins are reduced in the induced DCM hearts. This is observable at the ultra-structural level as well, with mitochondrial fractionation and thin, elongated sarcomeres readily detected by EM. Strikingly, many of the sarcomeric genes that are genetically downregulated in familial DCM patients were also reduced by IGF2BP2 expression in mouse hearts. Consistent with these observations, patients diagnosed with DCM or myocardial infarction have heightened levels of IGF2BP2, suggesting that our results in mice have clinical correlations and further strengthening the potential for therapeutic interventions by targeting the IGF2BP2 pathway.
In summary, we find that cardiac stress elevates the level of IGF2BP2, and induced IGF2BP2 leads to remodeling of the heart. This may be critical in the early stages of stress in order to compensate and maintain pumping under adverse conditions. A prolonged DCM phenotype can have deleterious effects, however, leading to heart failure and death. Reducing IGF2BP2 levels can rescue the heart, if lowered before there is irreversible damage. Having a big heart is not always a good thing.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in