A multiomics-based annotation of the herpes simplex virus genome
Published in Microbiology
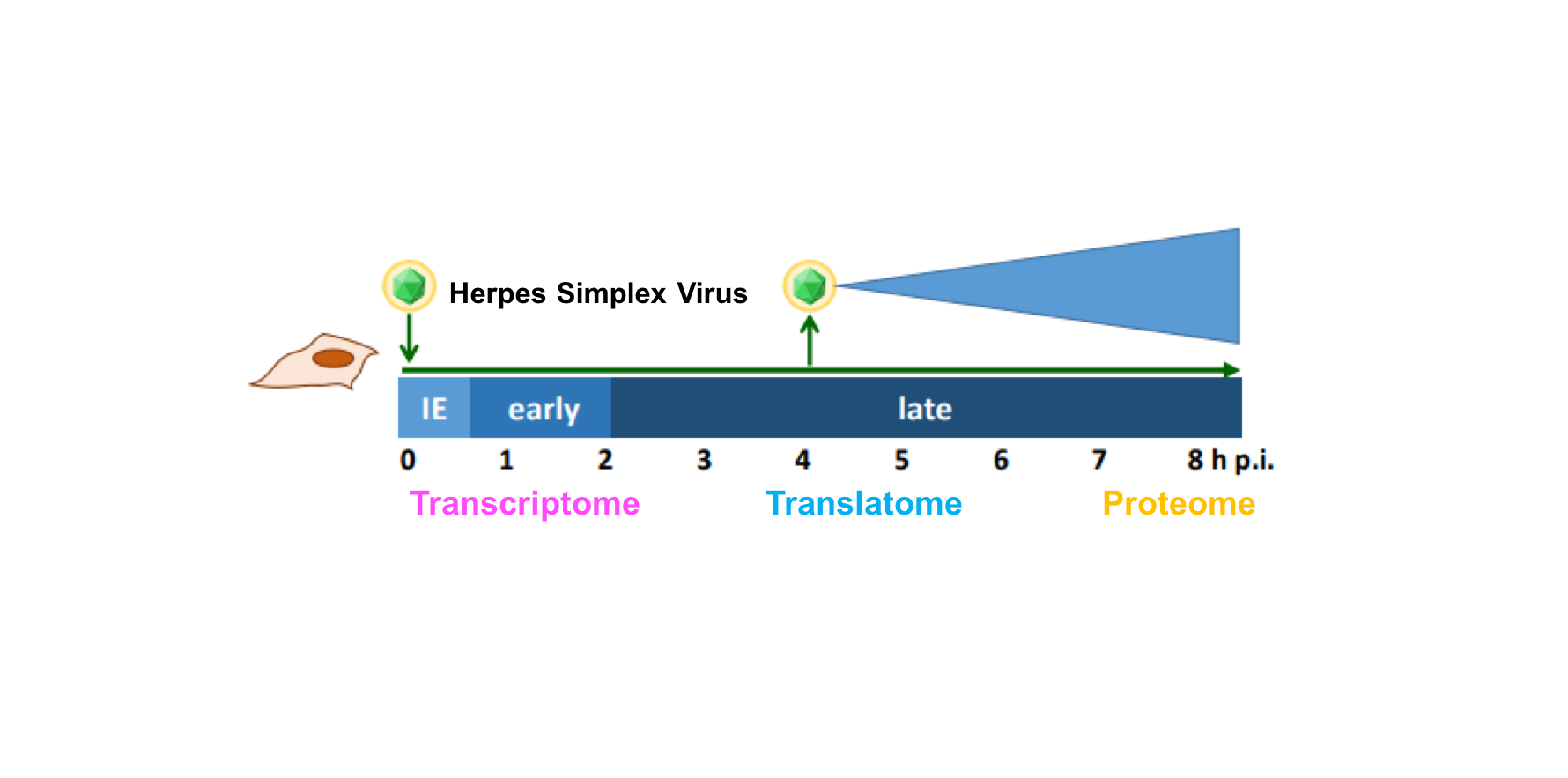
Though the ancient Greeks were the first to describe infections by herpes simplex virus (HSV), the cause of the common cold sore, an understanding of its basic biology was lacking until well into the 20th century. Over the past sixty years, HSV has been a valuable model to understand how viruses rapidly shut down host responses and reprogram cells into virus factories, while viral genome segments helped define broader topics in biology such as promoter regulation and DNA silencing. In addition to over half of the human population being naturally infected, HSV mutants with beneficial transgenes in place of virulence factors are prime candidates for oncolytic therapies and vaccine vectors, necessitating an understanding of how viral gene expression is regulated and how we can manipulate this to our advantage in the clinic.
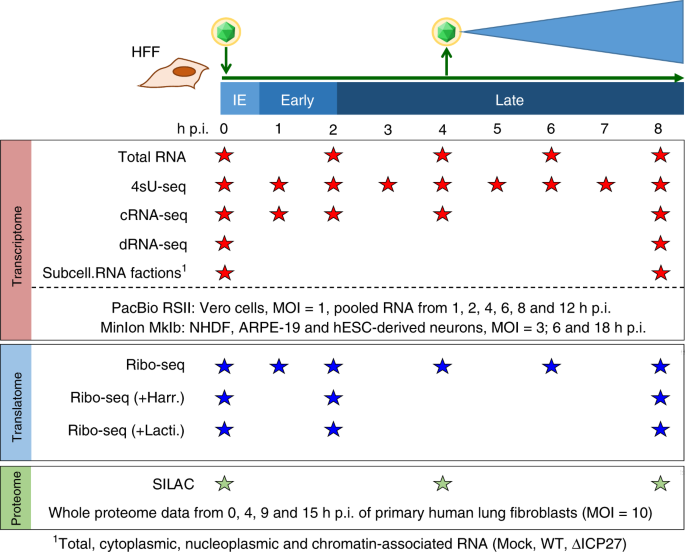
In our current work, we took a multi-faceted approach to determining the full repertoire of HSV gene expression throughout the course of lytic infection. By combining various kinds of RNA sequencing datasets from our lab with recent PacBio & MinION RNA-seq data, we mapped the precise locations of 201 viral transcripts with single-nucleotide precision. A major hurdle here was the combination of information from vastly different RNA-seq techniques to identify common transcription start sites (TiSS). For this task, we developed the tool iTiSS to automatically identify and rank TiSS found across different RNA-seq datasets while accounting for the advantages and limitations of each technique.
Before our work, it was widely accepted that HSV encodes roughly 80 proteins based upon a conservative definition of a coding sequence requiring at least 100 amino acids and beginning with an AUG start codon. By mapping the fragments of mRNA occupied by ribosomes (Ribo-seq), we identified over 200 additional open reading frames, many of which are much shorter than 100 amino acids and initiate with non-canonical start codons such as “CUG”. This flexibility in start codon usage, particularly late in infection when the cell’s physiology is most disrupted, enables temporally regulated expression of different protein isoforms. For two viral enzymes including the viral kinase Us3 in the figure below, isoforms initiating from upstream non-canonical start codons were found to have altered cellular localization during infection than their canonical, “AUG”-initiated counterparts and may thus serve different roles during infection.
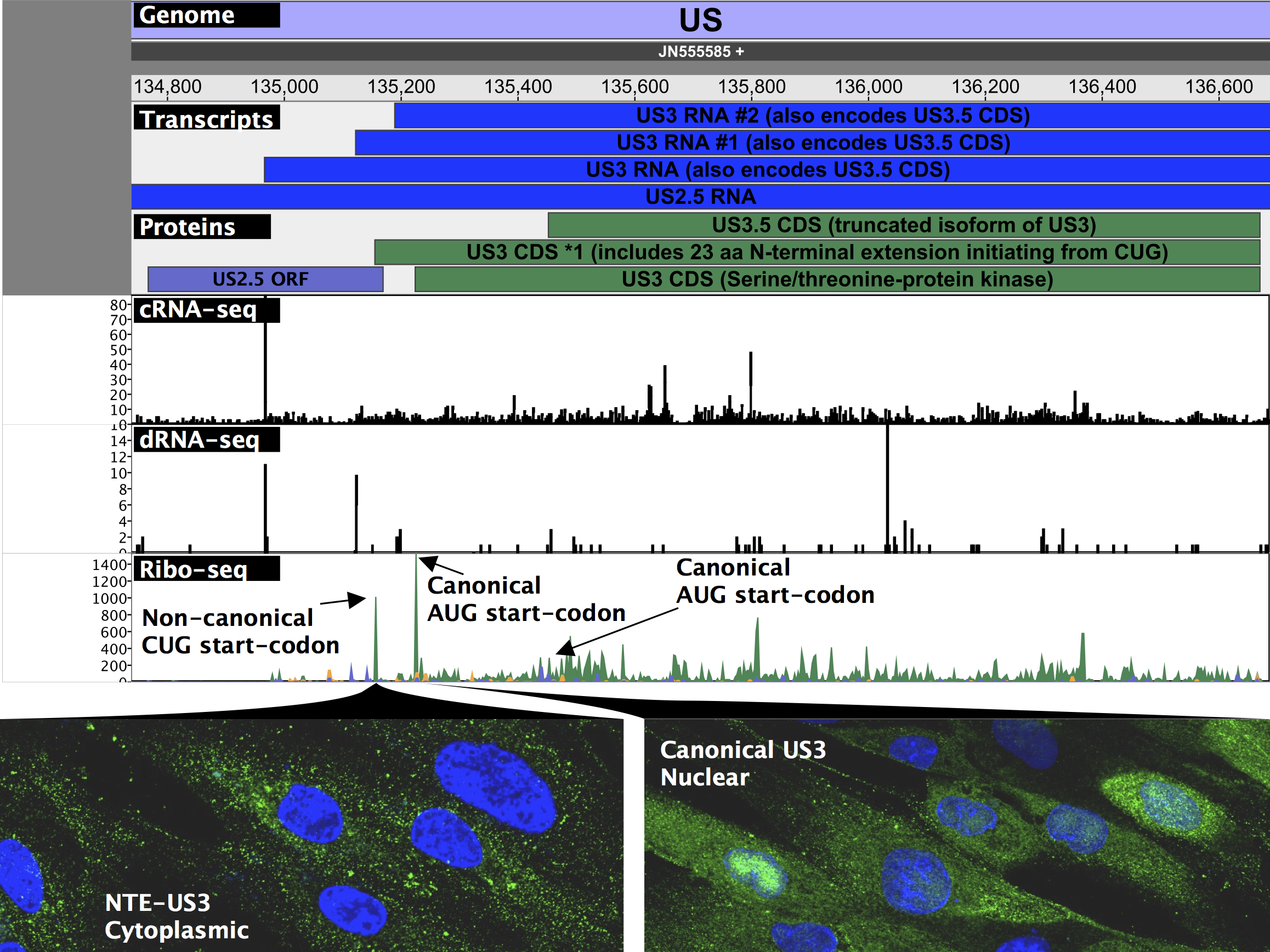
Naming all of the newly identified elements while maintaining the conventional nomenclature was a challenging –but necessary– task to make our data a widely usable resource. We have thus developed an extended annotation of both RNA and protein isoforms in herpesviral genomes that maintains existing gene names and leaves room for additional viral gene products and annotations in the future. We provide a genome viewer (shown above) that can be easily run on any operating system with an active Java installation that not only contains easy visualization of our extended annotation, but also of all data used in the course of this study. Furthermore, by providing access to our scripts and programs used to create figures and tables, executable with a single command along with detailed descriptions, we hope to do our part to help reproducibility and facilitate the integration of our data into future studies. We are excited to present our findings on the extended HSV coding capacity, and eager to see how our second-generation annotation improves over time along with our understanding of and ability to analyze herpesvirus gene expression.
Link to our study: https://doi.org/10.1038/s41467-020-15992-5
Link to our genome viewer and data: https://erhard-lab.de/software
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in