Active site remodelling of a cyclodipeptide synthase redefines substrate scope
Published in Chemistry

Cyclic dipeptides are composed of two amino acid building blocks, creating a structurally rigid 6 membered ring . Despite being the smallest of the cyclic peptide family, this class of molecules have been found to display remarkable properties such as blood-brain barrier permeability and resistance to bond hydrolysis, as well as being antibacterial, antifungal, anticancer and the list goes on. All of these factors have led to cyclic dipeptides being described as 'privileged' scaffolds making them attractive molecules for future research.
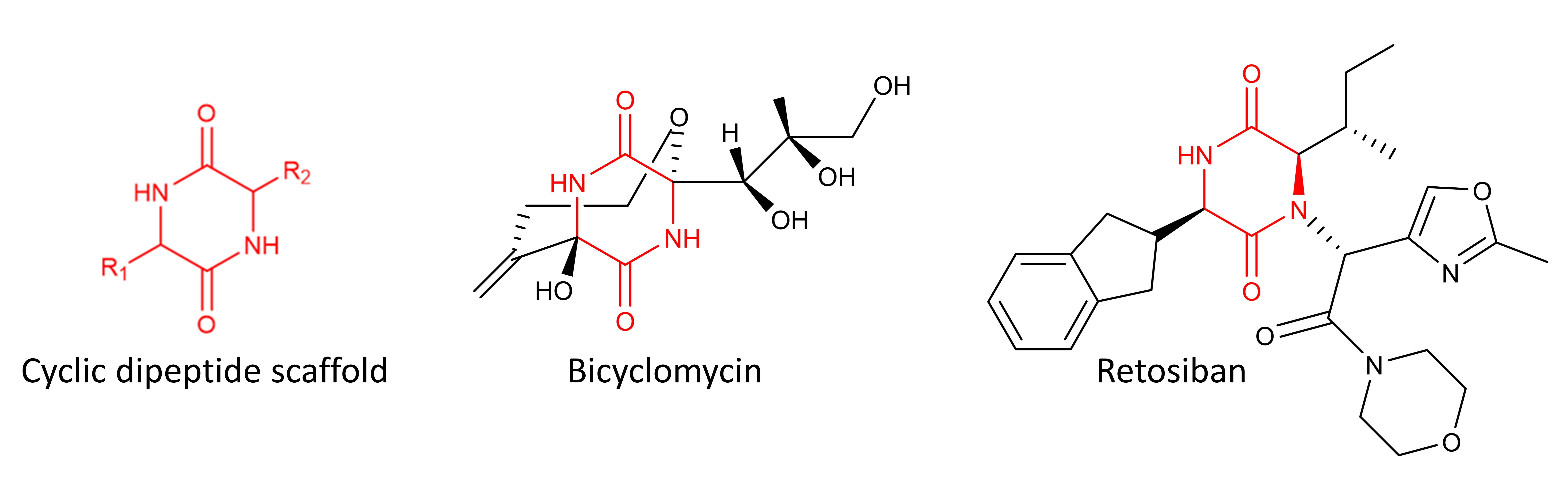 The common fold is shown in red and is featured in bicyclomycin and retosiban, both of which are current drug candidates on the market.
The common fold is shown in red and is featured in bicyclomycin and retosiban, both of which are current drug candidates on the market.
The synthesis of cyclic dipeptides was previously limited to conventional solution-based chemistry with long purification steps and little product yields. However, the discovery of biosynthetic pathways allowed us to harness the power of cyclodipeptide synthases (CDPSs), a family of enzymes capable of producing cyclic dipeptides. CDPSs repurpose the aminoacylated tRNA already present within the cell for use in cyclic dipeptide production.
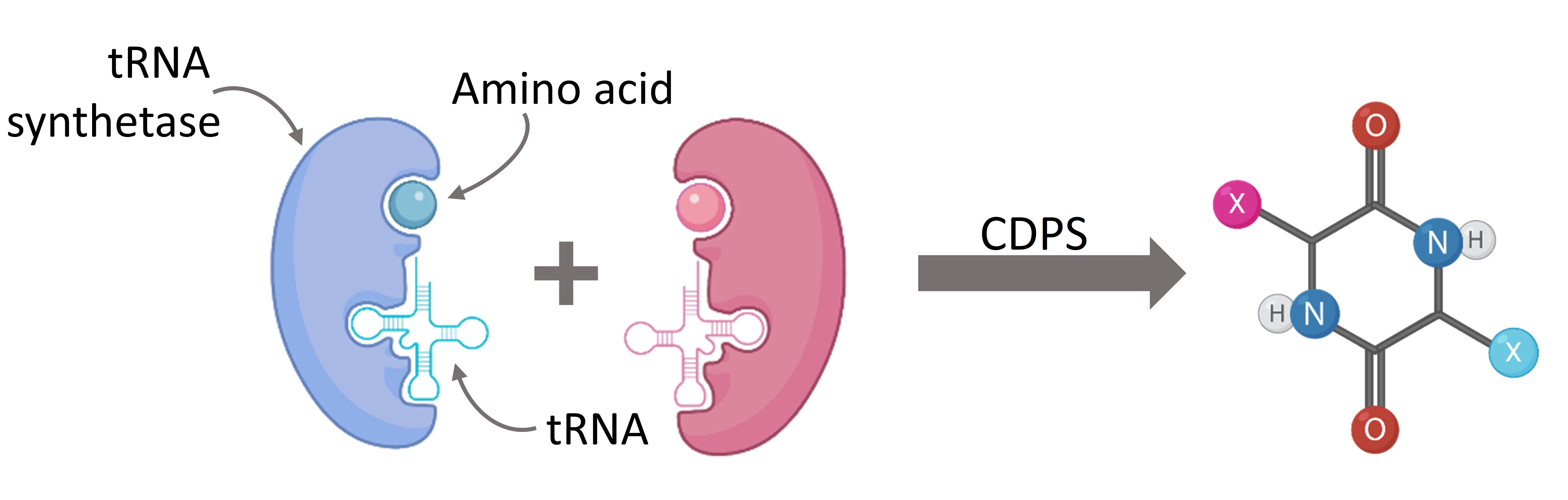
Our recently published work in Nature Communications Chemistry details an investigation into the only two known histidine-accepting CDPSs - ParaCDPS from Parabacteroides sp. 20_3 and ParcuCDPS from Parcubacteria bacterium RAAC4_OD1_1 which synthesise cyclo(His-Phe) and cyclo(His-Pro) respectively. Our work started with the optimisation of tRNA production to achieve a higher yield in a shorter time frame. Using our newly developed method containing all the tRNA present in E. coli, we investigated the promiscuity of the CDPS enzymes when unnatural amino acids were supplemented into the reaction. A small library of cyclic dipeptides was generated using two CDPSs; some of which have no synthetic method previously published. This finding led us to wonder how exactly ParcuCDPS was selecting for histidine as the first accepted amino acid in the reaction mechanism. The enzyme was crystallised and the structure was solved using heavy atom iodine phasing:
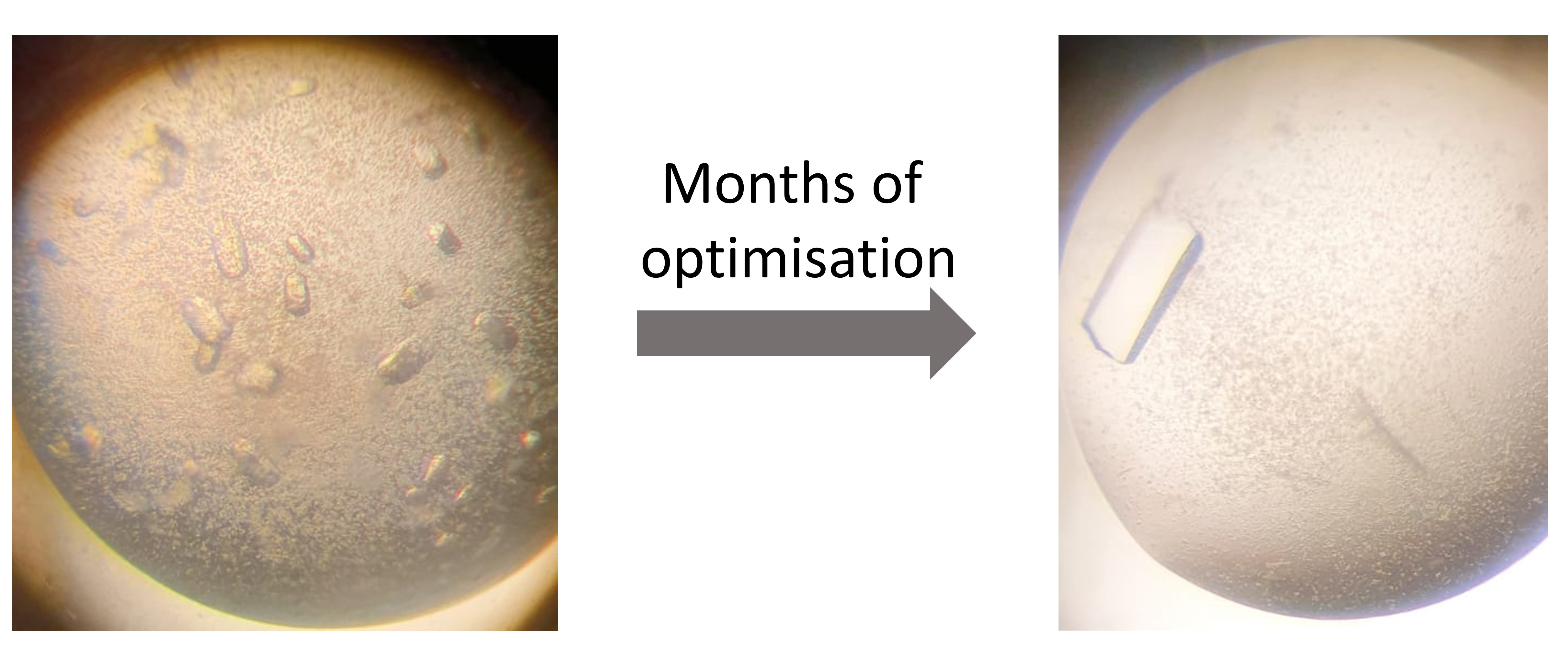
Having solved the structure of ParcuCDPS, we decided that wasn't enough and we created 24 mutants which were individually purified from E.coli, their activity tested and crystallisation trials conducted. In the end, 6 mutant structures were determined - all of which contained the same architecture as the original wild type structure.
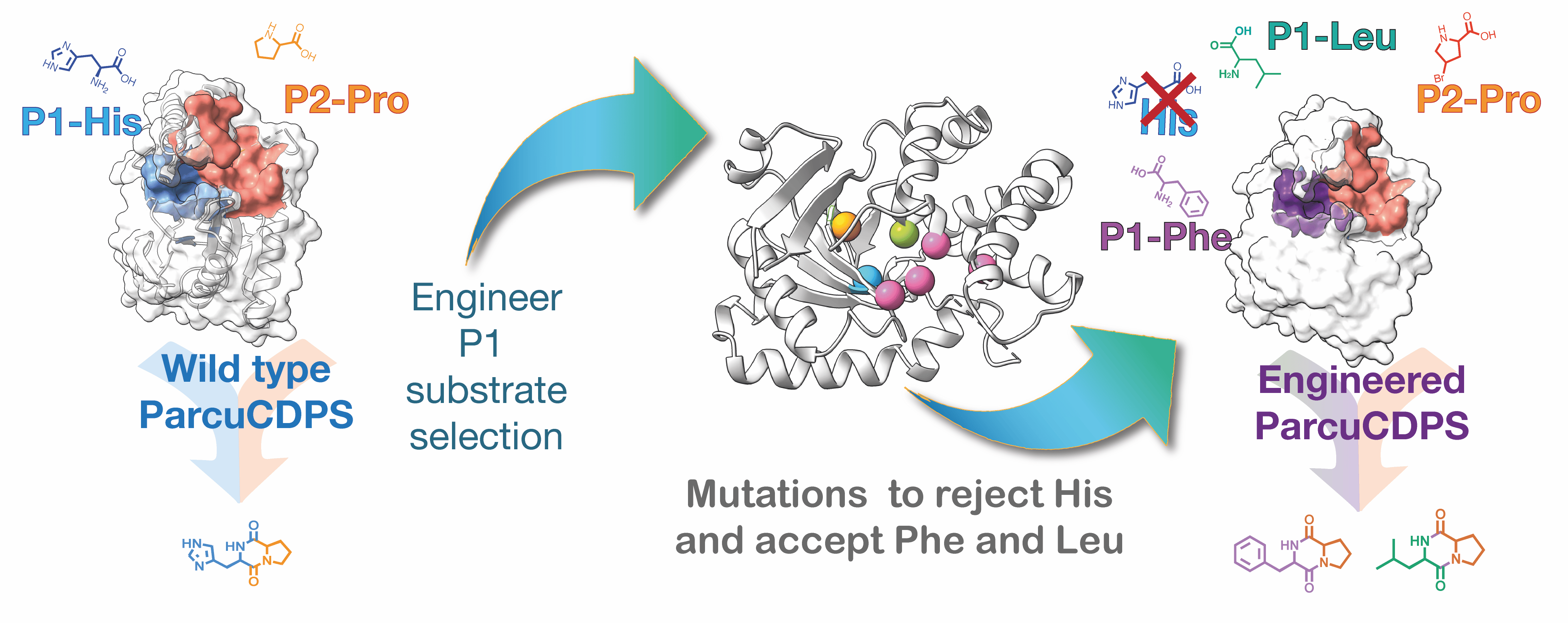
Lastly, we had this wild idea to investigate different amino acids with mutants that appeared inactive when using histidine. This experiment revealed that by changing three specific amino acid residues in the enzyme binding pocket, the substrate specificity of ParcuCDPS was shifted from histidine to phenylalanine or leucine. This is the first time that a CDPS enzyme has been successfully engineered to accept more than its original substrate.
For the full story, check out our article on Nature Communications Chemistry: Link to paper
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in