After the Paper | Highly-Accurate, Long-Read DNA Sequencing Improves Analysis of a Human Genome
Published in Bioengineering & Biotechnology

It has been very gratifying to follow the incredible work using highly accurate long-read sequencing, now commonly known as HiFi sequencing, since our publication on the topic in August 2019 (Nature Biotechnology 37, 1155-1162 (2019)). The pace of adoption and the creativity of the methods developed for analyzing HiFi reads certainly have surpassed our expectations.
Prior to HiFi sequencing, DNA sequencing technologies produced either high-accuracy short reads (e.g., Illumina, Ion Torrent) or lower accuracy long reads (e.g., PacBio, Oxford Nanopore). Historically, short reads excelled at identifying small variants in populations, and long reads at characterizing genome structure. We hypothesized that HiFi reads that were both long and accurate would provide better performance for both types of applications.
To test the concept, we sequenced a well characterized human sample, HG002 (Genome in a Bottle NIST Reference Material 8391), to high coverage with HiFi reads. We demonstrated that HiFi reads surpass standard short-read workflows for detecting single-nucleotide variants. Furthermore, HiFi reads beat noisy long reads in structural variant detection and the accuracy of de novo genome assemblies.
But challenges remained. HiFi reads did not perform as well as short reads for small (< 50 bp) indel calling. Software algorithms either treated HiFi reads as short reads (failing to utilize length) or as less accurate long reads (failing to utilize accuracy). The benchmarks used to measure performance did not adequately include the new regions of the genome covered by HiFi reads. Most importantly, it was expensive, complicated, and laborious to generate HiFi reads. Sequencing HG002 required nearly two months of a DNA sequencing instrument, 120,000 CPU core-hours to generate consensus reads, and dedicated efforts of tens of scientists.
The last year has brought progress in all those areas. The PacBio Sequel IIe System produces 10-fold more HiFi reads than the instrument used in our study for the same amount of time and cost. The software for generating HiFi reads has been sped up by around 50-fold and then ported to run on the Sequel IIe System, eliminating cost and complexity. The Genome in a Bottle Consortium adopted HiFi reads to improve its benchmarks to cover an additional 7% of the human genome.
Software tool developers rapidly embraced HiFi reads, designing algorithms that utilize both the accuracy and long-range information. With these algorithms, HiFi reads now match short reads for indel calling and produce reference-quality genome assemblies in hours.
The vast majority of PacBio sequencing is now HiFi-based. HiFi reads have moved long-read sequencing from niche applications to providing the best quality for genome assembly and comprehensive variant detection. Highlights of this progress from the research community are:
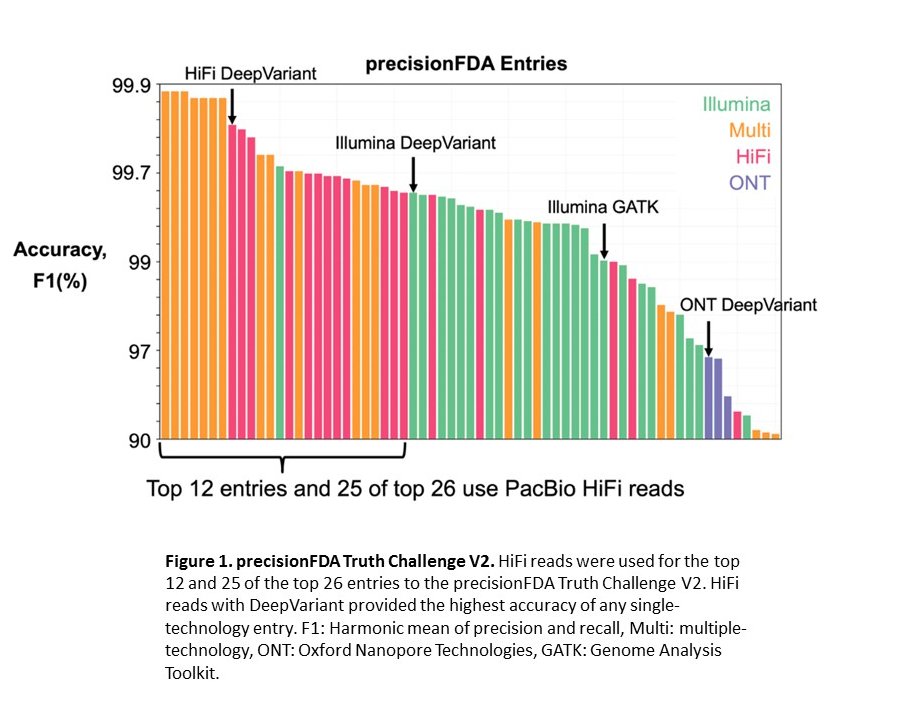
Google DeepVariant with HiFi reads wins precisionFDA Truth Challenge V2
Our initial work used DeepVariant v0.7 software to call variants in the human genome. Performance matched short reads for single nucleotide variants but underperformed for indels. Since then, DeepVariant has been trained on improved truth sets and been extended to utilize phasing information in HiFi reads and expose more read information by re-aligning to candidate variants. The current DeepVariant v1.0 has 80% fewer errors than the v0.7 software for the same HiFi reads.
HiFi reads with DeepVariant now provide the best accuracy for small variant calling of any single technology, as demonstrated by the precisionFDA Truth Challenge V2, which evaluated methods for variant calling in human genomes (Figure 1).
HiFi sequencing solves rare disease cases undiagnosed by other approaches
The technical superiority of HiFi sequencing for variant calling inspired efforts to apply it to rare disease cases for which short-read sequencing did not identify a pathogenic variant. One study led by Gregory Cooper applied HiFi sequencing to six unsolved cases with neurodevelopmental disorders. Strong candidate variants were identified in two cases: a “likely pathogenic (LP), de novo L1-mediated insertion in CDKL5 that results in duplication of exon 3, leading to a frameshift” in one proband, and “translocations affecting DGKB and MLLT3” in another.
A separate study led by Mary-Claire King evaluated variation in BRCA1 in families with a history of early-onset breast cancer but no previously detected pathogenic variants. For one family, “severely affected with early-onset bilateral breast cancer and with negative (normal) results by gene panel and exome sequencing”, HiFi reads identified “an intronic SINE-VNTR-Alu retrotransposon insertion that led to the creation of a pseudo exon in the BRCA1 message and introduced a premature truncation.”
The success of these studies has inspired much larger ongoing studies from groups like Invitae and Children’s Mercy Hospital, Kansas City.
Telomere-to-Telomere genome assemblies with HiFi reads
We discussed in our publication how the accuracy of HiFi reads resolves similar but non-identical repeats without needing to fully span the repeats. The HiCanu and hifiasm assemblers apply this concept – including multiple improvements to squeeze more accuracy from HiFi reads – to generate reference-quality assemblies from HiFi reads alone.
A HiCanu assembly served as the base for the Telomere-to-Telomere (T2T) Consortium to produce the first ever complete gap-less assemblies of most chromosomes for one human individual. The Human Pangenome Reference Consortium is using HiFi reads and similar assembly approaches for hundreds of human individuals with the goal of updating the human reference genome to better represent the entire population.
HiFi sequencing of Earth’s biodiversity
HiFi reads have also been applied successfully to assemble genomes larger and more complicated than human, including oat, rose, and redwood (a 27 Gb hexaploid). On the other end of the size spectrum, HiFi sequencing has opened opportunities to sequence metagenomes and smaller species with little available DNA that previously were extremely challenging for long reads. To support further methods development and allow interested scientists a chance to evaluate the power of HiFi data, we have recently published a paper describing five publicly available HiFi datasets covering a diverse set of organisms. The data set spans a range of genome sizes, complexities, and ploidy levels: including inbred maize, an inbred mouse, an outbred diploid frog (17 Gb genome), an octoploid strawberry, and a mock metagenome community.
I am confident that there remains much headroom to improve HiFi sequencing and its applications, and I am excited to see what progress will come in 2021 and beyond!
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in