An insightful journey: unraveling the cellular impact of an antitumoral drug to reveal its broad antiviral activity
Published in Microbiology, Cell & Molecular Biology, and Biomedical Research
Right at the beginning of the COVID-19 outbreak in 2020, researchers and pharmaceutical industries worldwide joined forces to develop vaccines while we were rapidly gaining knowledge of the new virus that caused the emergency. Although vaccines were developed in the shortest time ever recorded, the pandemic had already resulted in the loss of millions of lives. This clearly illustrates why having effective antivirals in place when new pandemics begin could be a gamechanger in future outbreaks.
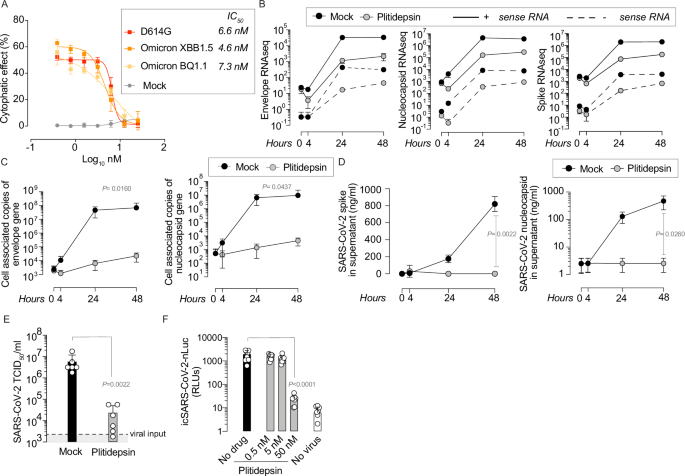
In 2020, a research team was launched in Barcelona at the IrsiCaixa research institute. This new group called PISTA was devoted to the study of emergent pathogens, immunity, signaling and therapeutic applications. Together with other researchers from the CBIG consortium, they worked at high biosafety laboratories exploring clinically approved drugs to find antivirals against SARS-CoV-2. During this screening performed in the early days of the pandemic, an antitumoral drug named plitidepsin and used for treating myeloid leukemia stood up and was effective inhibiting SARS-CoV-2 replication at nanomolar concentrations (1).
Shortly after, I joined the PISTA team to begin my PhD thesis on antiviral therapies, and plitidepsin became my mystery to solve. What we already knew is that the molecular target of plitidepsin is EEF1A, a protein needed for the elongation of the polypeptide chain during protein translation (2). Thus, we were in front of a host-directed drug that targets an essential pathway for the cell that was also effective against SARS-CoV-2 replication. Therapeutic targeting of the host rather than the pathogen offers many key advantages, including a higher potential for broad-spectrum antiviral activity that may be especially useful in an era when various viral threats may emerge. However, these host-directed drugs may present potential toxicity to our cells, as they target key cellular pathways necessary for survival. Thus, this became our first question to address. Many days passed checking under the microscope observing how the cells reacted to the treatment at the concentrations at which the drug exerts antiviral activity. We used different methods to analyze their viability, and impressively enough, they were always alive and not infected. It then became a challenge to find out what was happening inside those cells that survived while a crucial mechanism like protein translation was blocked, and the replication of SARS-CoV-2 was halted.
At this point, we embarked on a long journey to explore how the cells were responding to the treatment on a broader level, by conducting experiments focused on transcriptomics, proteomics and functional assays transfecting different RNA reporters, all with the same objective: unraveling the cellular landscape after plitidepsin treatment beyond its known mechanism of action on the molecular target EEF1A.
This journey was not only scientific but also deeply personal, as each new experiment brought a new set of questions, which required an open mind and patience to explore and understand what results were revealing. I will never forget the experiment in which I transfected my cells with a dual reporter plasmid coding via cap-dependent or IRES-dependent translation, expecting that IRES translation would not be blocked by plitidepsin. Results showed the complete opposite, what led to my initial frustration. However, after careful evaluation of all the pieces of the puzzle, we discovered an alternative mechanism of protein translation, named m6A-pathway, that opened a new path in our journey to understand our data. Personally, this journey felt like slowly pulling a thread from a tangled network of proteins upregulated by plitidepsin, that initially appeared disconnected and meaningless but slowly started to make sense after spending days reading bibliography and performing functional assays with patience and dedication. This process is always easier in a team where people work together with resilience and personal support, while cooperating to bring new ideas.
This is how we ended up showing that, after blocking an essential cellular pathway such as protein translation with an antitumoral drug, the cells generated a molecular fingerprint characterized by the upregulation of alternative translation initiation factors of the m6A-pathway. This particular route of translation not only led to cellular survival in the absence of SARS-CoV-2 replication but also allowed us to predict and demonstrate in vitro the effect of the drug against different viral families. These viruses use cap-dependent and IRES-dependent pathways to replicate, including MERS-CoV, Zika, HSV, HCVr and RSV (Figure 1). However, for viruses using the alternative m6A-pathway, as is the case of HIV-1, plitidepsin did not block replication (Figure 1). Understanding the molecular fingerprint of a therapy provides a broader perspective that goes beyond its mechanism of action. This is crucial to explore the utility of host directed therapies as broad-spectrum antivirals for emerging viral threats.

- Rodon, J. et al. Identification of Plitidepsin as Potent Inhibitor of SARS-CoV-2-975 Induced Cytopathic Effect After a Drug Repurposing Screen. Front. Pharmacol. 12, 976 646676 (2021).
- Losada, A. et al. Translation Elongation Factor eEF1A2 is a Novel Anticancer Target for the Marine Natural Product Plitidepsin. Sci Rep 6, 35100 (2016).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in