
From Ancient Greece to the Renaissance and beyond, symmetry has fascinated artists and scientists alike. While symmetrical forms of matter have been the focus of intense research efforts, strategies to disrupt symmetry at a molecular level still represent a major challenge today. As a representative example, the asymmetric construction of all-carbon quaternary centers, motifs present in a wide variety of biologically active molecules, is still far from being considered a solved task. Further, their incorporation in acyclic systems is even more demanding due to the additional degrees of freedom and the steric congestion at the stereogenic center.
Rearrangement reactions represent a powerful strategy towards this end as they can take advantage of cyclic intermediates to transfer stereochemical information from the starting material into the final product. In recent years, the Smiles rearrangement has undergone a true revival for the synthesis of valuable building blocks. In its Smiles-Truce variant, an intramolecular nucleophilic aromatic substitution reaction takes place, either under strong basic or radical conditions, involving the concomitant cleavage of a Csp2-X bond and formation of a new Csp2-C bond by ipso substitution via a spirocyclic intermediate (Fig. 1, top).
In this context, our group has recently developed a series of one-pot radical Smiles rearrangement cascade reactions, triggered by radical additions to π-systems. Following addition of a radical to N-arylsufonyl acrylamides, a variety of α-aryl-β-substituted amides and indoles containing all-carbon quaternary centers were accessed in a highly efficient manner (Fig. 1, bottom).
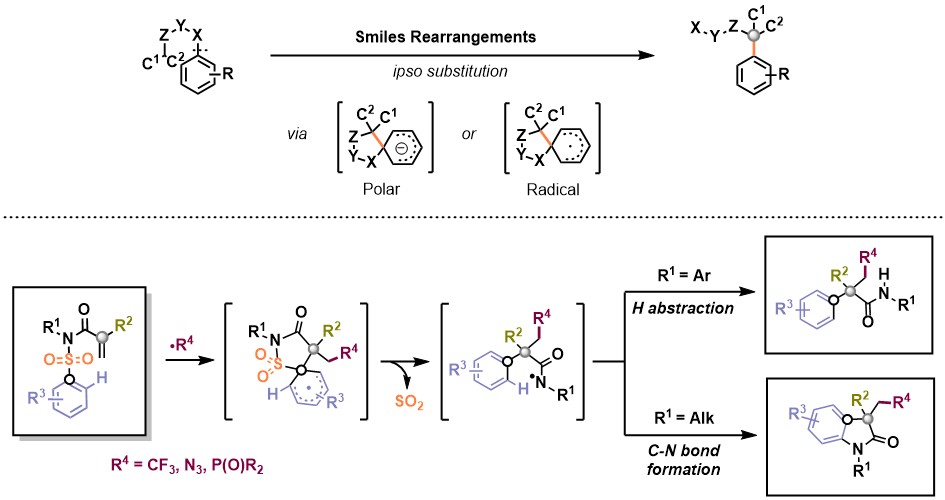
Fig. 1. Smiles-Truce rearrangement
Inspired by the potential of radical Smiles rearrangements to meet the challenge of generating quaternary stereocenters, we sought to develop an asymmetric version of this transformation. We hypothesized that the presence of a chiral linker conjugated to the aromatic migrating group could induce enantioselectivity in this process. Sulfoxides were envisioned as “traceless” chiral auxiliars based on their recognized ability to impart stereocontrol in a broad variety of organic reactions. Further, we hypothesized they could be released in situ under the reaction conditions. Finally, photoredox catalysis would enable the facile generation of radicals, significantly expanding the scope of this transformation (Fig. 2).
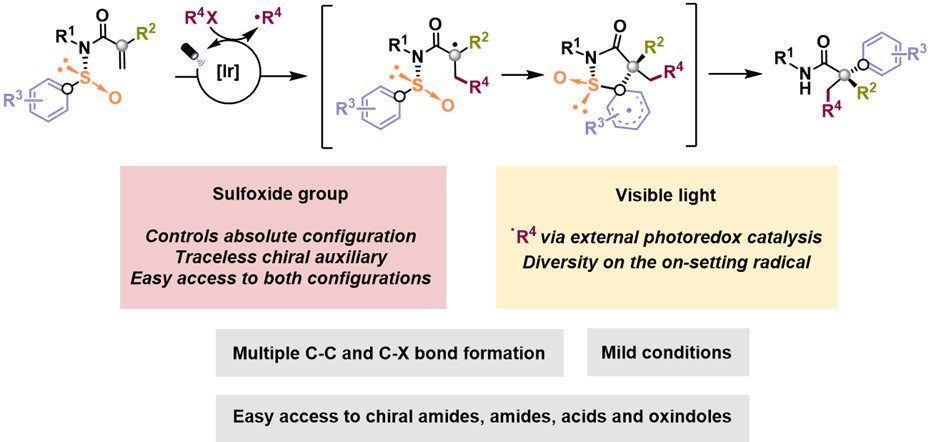
Fig. 2. Our strategy for the enantioselective synthesis of all-carbon quaternary centers in acyclic systems.
In our recent work published in Nature Chemistry (2021, VOL 13 | April 2021 | 327–334), we used enantioenriched N-arylsulfinyl acrylamides as acceptors for a variety of C- and S-based radicals, produced in situ under mild photoredox conditions. The sulfoxide group in the starting material not only directs the 1,4-migration of the aryl moiety onto the α-carbon of the amide, thus governing its absolute configuration, but also acts as a traceless chiral auxiliary. The α-all-carbon substituted amides obtained in this multicomponent process are prevalent in pharmaceuticals, agrochemicals, and bioactive natural products. Further, they can be transformed, in a single additional step, into valuable chiral building blocks difficult to obtain by other methods (e.g. α,α-disubstituted acids, oxindoles and β,β-disubstituted amines), thus highlighting the overall synthetic potential of our method (Fig. 3).
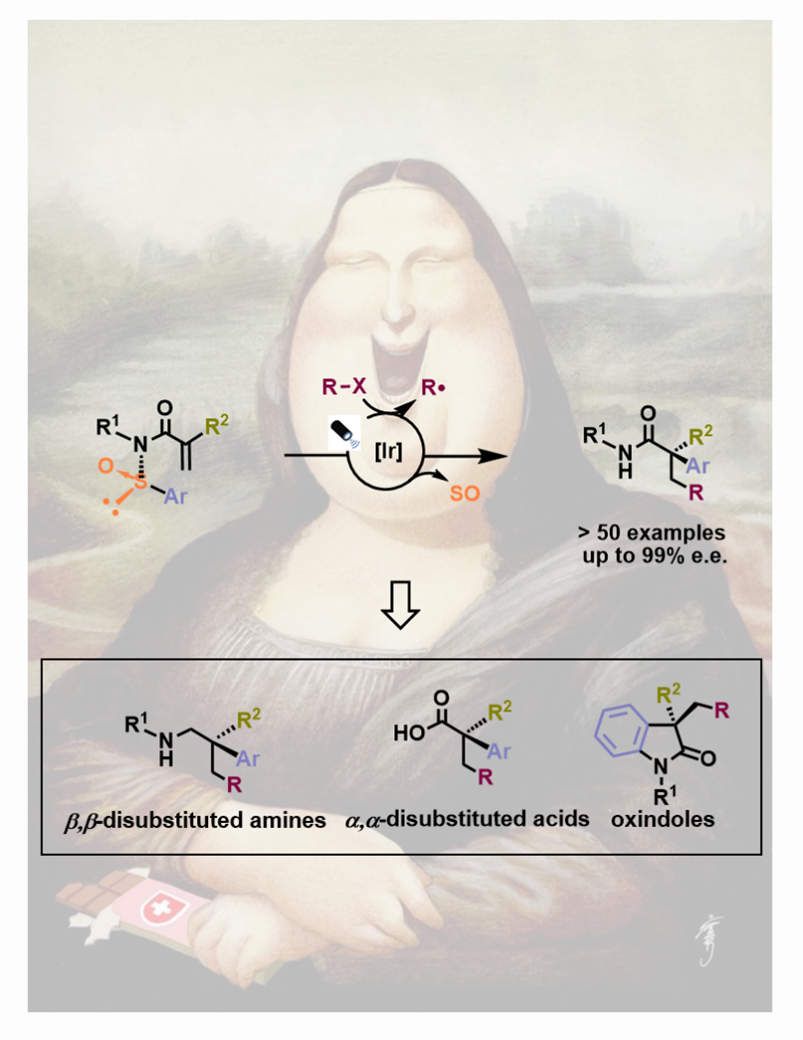
Fig. 3. Access to valuable chiral building blocks
We believe that these investigations mark a milestone for obtaining all-C quaternary centers in acyclic systems and open a new avenue of exploration on the formation of these motifs in a broad diversity of chemical structures.
Follow the Topic
-
Nature Chemistry

A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in