BID-seq: A quantitative method mapping pseudouridine (Ψ) sites at base resolution
Published in Bioengineering & Biotechnology

Since our laboratory first proposed RNA modifications in broad gene expression regulation in 2010[i], most studies in the RNA epi-transcriptome field have been concentrated on m6A in mRNA, while Ψ has been studied much less despite that its level in mammalian mRNA is almost as high as that of m6A. There are thirteen annotated pseudouridine synthetases (PUS) encoded in the human genome; most of these proteins deposit Ψ to different RNA species including mRNA, rRNA, tRNA, snRNA, etc[ii]. Mutations in these PUS enzymes have been associated with different human diseases[iii]. While pseudouridylation in abundant RNA species such as rRNA and tRNA has been well studied, the distribution and functions of Ψ in low abundant RNA species like mRNA remain unclear, mostly due to the lack of a base-resolution method that can reveal the exact Ψ modification stoichiometry at modified site.
N-cyclohexyl-N’-(2-morpholino ethyl) carbodiimide methyl-p-toluenesulfonate (CMC) is a chemical known to react with Ψ to form CMC-Ψ adduct, which can induce RT truncation signatures[iv]. Two reports using this principle to sequence Ψ in mRNA were published in 2014[v],[vi]. In these CMC-based methods, high modification fraction at a Ψ site is typically required to confidently read out the RT truncation signature above background noises, as many other factors may also cause RT stop. To increase sensitivity, an azide-modified CMC was developed later to enrich Ψ-containing RNA fragments for sequencing, allowing detection more Ψ sites[vii]. However, it lacks base-resolution and stoichiometry information at the modified sites. These CMC-based methods stimulated the research in Ψ field, but functional studies on Ψ are still hampered by the lack of a quantitative sequencing method for Ψ.
In 2019, a PNAS paper reported the application of bisulfite treatment to sequence Ψ. When conducting conventional bisulfite sequencing of m5C in tRNA, Khoddami et al observed that the known Ψ sites in tRNA generated a unique deletion signature as a new principle for Ψ detection (Fig. 1A)[viii]. They next developedRBS-seq to detect Ψ sites in rRNA and mRNA as deletions. However, RBS-seq used the conventional BS conditions under acidic pH, with limited Ψ sites detected in mRNA and undesired C-to-U conversion occurring at the same time. Based on the understanding of BS sequencing mechanism, we thought that these drawbacks could be overcome by changing the pH of BS treatment from acidic to neutral. Since the reaction U with BS is optimal under neutral conditions, the reaction of Ψ with BS may be optimal at neutral as well. To test the idea, we synthesized short RNA oligos containing either Ψ or C and screened BS reaction conditions including different pH, ion concentration, incubation temperature, and reaction time, using mass spectrometry to monitor the reactions. We characterized the optimized BS conditions that quantitatively converted Ψ to Ψ-BS adduct without any detectable C-to-U conversion. We then further screened RT enzymes and found SSIV generated a high deletion rate at the fully modified Ψ site and meanwhile close to 0% deletion ratio in untreated input. We later established BID-seq pipeline (Fig. 1B) and built libraries with RNA oligos containing NNΨNN (N = A or C or G or U) and found that 231 out of 256 motifs gave deletion rates over 50% at Ψ site, allowing us to quantify Ψ modification fraction (Fig. 2A). We further validated BID-seq by detecting all the known Ψ sites in human rRNA and quantified the Ψ fraction at each modified site. We applied BID-seq and uncovered thousands of Ψ sites in mRNA from human cell lines and mouse tissues (Fig. 2B and 2C), providing the first comprehensive map of Ψ with quantitative information and single-base resolution.
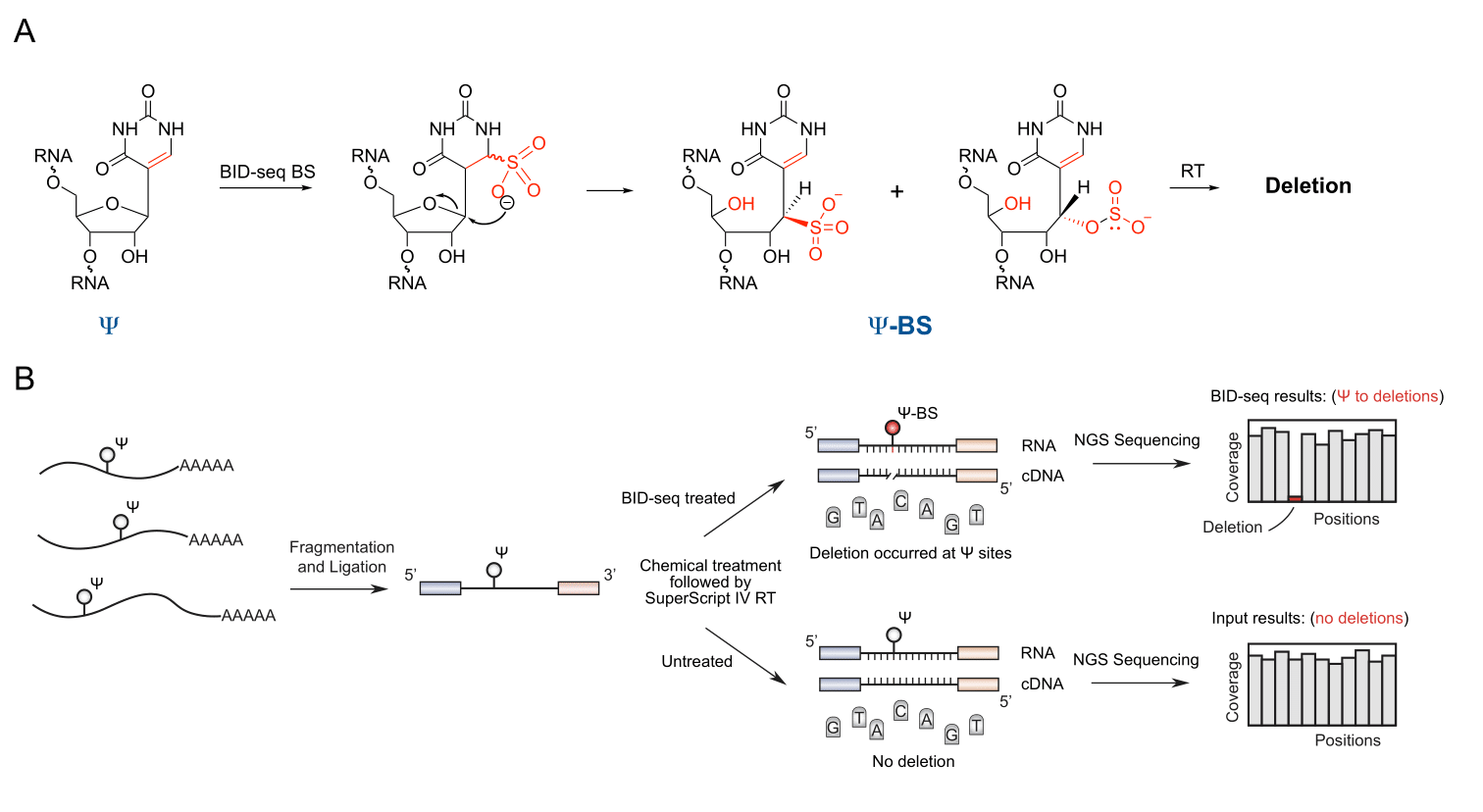
Fig. 1 | The development of BID-seq to map Ψ as deletion signatures.
(A) The chemical structure of the Ψ-BS adduct after bisulfite treatment, which induces deletion signatures during reverse transcription. (B) A flowchart of library construction pipeline for BID-seq, revealing Ψ modification fraction by deletion ratio signature.
The quantitative nature of BID-seq allowed us to assign specific pseudouridine synthase to individual Ψ sites in human mRNA. We were able to assign ‘writer’ protein to hundreds of frequently modified Ψ sites and found the presence of highly-modified Ψ stabilizes mRNA. We found that mouse tissues contain a lot more highly modified Ψ sites on mRNA than cell lines (Fig. 2B), suggesting potential broad roles of mRNA Ψ in tissue or primary cells. For the first time, BID-seq provided the in vivo evidence that Ψ exists in stop codons of many mRNAs from human and mouse, although it has been known for a long time that the replacement of U by Ψ can convert stop codons to sense codons in vitro. We observed at least 106 Ψ-modified stop codons from 12 mouse tissues, suggesting stop codon read-through via pseudouridylation as a potential mechanism to modulate translation in mammals. Taken together, BID-seq will enable future investigations of the roles of Ψin diverse biological processes, and impact life science research.
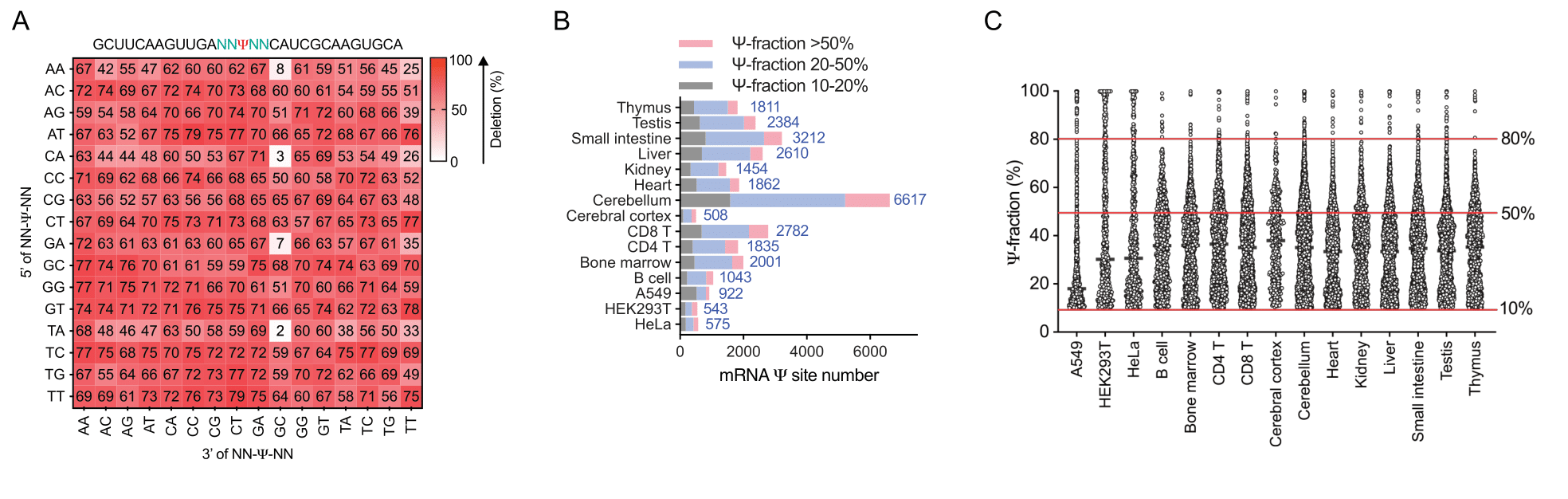
Fig. 2 | BID-seq uncovers abundant Ψ in mRNA from human cell lines and mouse tissues, highlighting numerous highly modified Ψ sites.
(A) Heatmap plot for deletion ratios on 256 motifs (NNΨNN) after bisulfite treatment in BID-seq, which contain one 100% modified Ψ within each motif. (B) BID-seq reveals a large number of Ψ sites (modification fraction above 10%) in 12 mouse tissues and 3 human cell lines. (C) The modification level distribution of mRNA Ψ sites in 12 mouse tissues and 3 human cell lines, in which numerous Ψ sites are highly modified (modification fractions above 50%).
[i] He, C. Grand challenge commentary: RNA epigenetics? Nat. Chem. Biol. 6, 863-5 (2010)
[ii] Rintala-Dempsey, A.C. & Kothe, U. Eukaryotic stand-alone pseudouridine synthases–RNA modifying enzymes and emerging regulators of gene expression? RNA Biol. 14, 1185–1196 (2017).
[iii] de Brouwer, A.P.M. et al. Variants in PUS7 Cause Intellectual Disability with Speech Delay, Microcephaly, Short Stature, and Aggressive Behavior. Am J Hum Genet 103, 1045–1052 (2018).
[iv] Bakin, A. & Ofengand, J. Four newly located pseudouridylate residues in Escherichia coli 23S ribosomal RNA are all at the peptidyltransferase center: analysis by the application of a new sequencing technique. Biochemistry 32, 9754–9762 (1993).
[v] Carlile, T.M. et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143–146 (2014).
[vi] Schwartz, S. et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159, 148–162 (2014)
[vii] Li, X. et al. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 11, 592–597 (2015).
[viii] Khoddami, V. et al. Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution. Proc. Natl. Acad. Sci. USA 116, 6784–6789 (2019).
Trained as a Ph.D. in organic chemistry, I have been working on nucleic acid chemistry and synthesized many DNA and RNA oligos containing various modifications as models to study biochemistry. Since 2012, a K01 award from NIH allowed me to extend my research to chemical biology, mainly focusing on developing new sequencing methods for various DNA and RNA modifications.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in