CD8+ T cell metabolic flexibility elicited by CD28-ARS2 axis-driven alternative splicing of PKM supports antitumor immunity
Published in Immunology
Background:
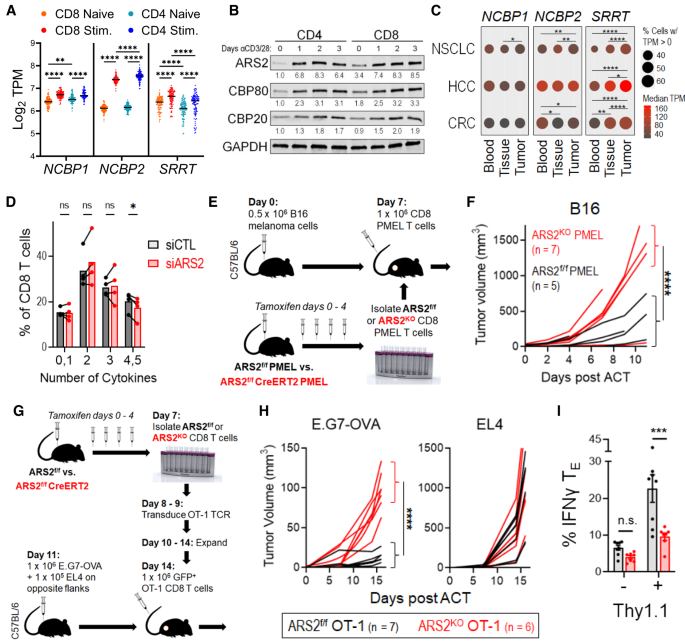
This study initially stemmed from our previous observations regarding the role of ARS2 in hematopoiesis and T cell development (thymopoiesis; Elahi 2018). As T cells progress through thymopoiesis, one of the key developmental stages involves selection for T cells that express a functional T cell receptor (TCR), engagement of which drives robust proliferation. When we developed a T cell specific ARS2 knockout mouse (ARS2f/f Lck-Cre), we found loss of ARS2 resulted in a profound increase in cell death specifically at this stage of thymopoiesis. However, T cells isolated from the spleens of these mice showed normal levels of ARS2 expression, suggesting essentially all ARS2KOthymocytes apoptosed and failed to reach the periphery. Considering our previous findings showing ARS2 is upregulated upon mitogenic stimulation, this led us to wonder whether ARS2 might undergo similar regulation in mature T cells.
Publicly available data demonstrated ARS2 mRNA (SRRT) was increased in both CD4+ and CD8+ human T cells following stimulation, and was specifically elevated in tumor infiltrating CD8+ T cells. We confirmed these findings, and further found ARS2 induction to be dependent on CD28 costimulation. Around this time, I was presenting my work at our joint lab meeting with Kelvin Lee’s lab when he pointed out that they had several mouse lines with knock-in mutations in the CD28 intracellular signaling domains to render them non-functional: the membrane proximal YMNM to which PI3K binds (mutated to Y170F), the distal PYAP which is bound by Grb2 and Lck (AYAA), or both (DKI – double knock-in), as well as a full CD28 knockout. When we stimulated T cells from these mice we observed that PI3K signaling was completely dispensable to ARS2 upregulation; instead, the PYAP motif was required. When Jonathan Green had originally developed these mice, he found the PYAP motif to be essential to in vivo effector function, leading us to speculate that upregulation of ARS2 downstream of Grb2/Lck could provide a means for cells to coordinate changes in gene expression with changes in cell state (such as the differentiation from a naïve to effector phenotype). In agreement with our hypothesis, ARS2KO and CD28AYAA, but not CD28Y170F CD8+ T cells were defective in their ability to control antigen-specific tumor growth in vivo and produce inflammatory cytokines (IL-2, TNFα, and IFNγ).
To understand how ARS2 affects T cell function following stimulation, we then carried out RNA-seq of WT, ARS2KO, and CD28 mutant T cells. Surprisingly, ARS2 deletion had relatively minor effects on differentially expressed genes. However, we knew from early studies of CD28 function carried out in the 1990s by Craig Thompson and Carl June that CD28 also exerts important effects on RNA biology, such as mRNA stability. More recently, Kristen Lynch and Arlene Sharpe had shown T cell activation was accompanied by global changes in alternative splicing, and that this occurred in a CD28-dependent manner, respectively. We therefore conducted PSI-Sigma splicing analysis and found ARS2 to be required for ~1/3 of activation-induced splicing events 3 days post-activation. Among genes that underwent CD28-PYAP and ARS2-dependent splicing, we identified the glycolytic enzyme pyruvate kinase (Pkm), which was spliced to the M2 isoform (Pkm2) upon activation. Mechanistically, we showed ARS2 interacts with Pkm pre-mRNA and recruits the splicing factors SRSF3, hnRNPA1, and PTBP1 to facilitate Pkm splicing, thus providing the first direct evidence that ARS2 regulates alternative splicing.
PKM is the final rate-limiting enzyme in glycolysis, and has also been proposed to have a non-metabolic ‘moonlighting’ function in the nucleus, though this notion remains controversial as other studies (Hosios 2015, Johnson 2023) have shown PKM2 does not have any serine/threonine protein kinase function. Functionally, PKM2 shows a slower rate constant than PKM1 and, in the context of tumors, this is thought to allow for the accumulation of upstream glycolytic intermediates that can be shuttled into anabolic pathways in support of rapid proliferation. Crucially, in T cells, glycolytic flux also regulates effector cytokine production – when cells are grown in the absence of glucose GAPDH binds cytokine mRNA (e.g. Ifng) and prevents translation. However, the buildup of glycolytic intermediates inhibits GAPDH binding and allows for cytokine protein synthesis. Thus, it stands to reason that PKM2, and the reduced flux of glucose carbons into pyruvate, would result in the accumulation of upstream intermediates and allow for IFNγ production. Consistent with this, cells with a reduced ratio of PKM2:PKM1 (ARS2KO, CD28AYAA, and PKM2KO) all exhibited decreased glycolytic metabolism accompanied by reduced IFNγ and IL-2 protein production, despite expressing equivalent levels of cytokine mRNA.
In summary, our data suggest a model where ARS2 upregulation downstream of CD28 costimulation facilitates Pkm alternative splicing. Increasing the ratio of PKM2:PKM1 confers T cells with the metabolic flexibility necessary to carry out effector functions in the metabolically hostile tumor microenvironment. It will be intriguing for future studies to assess the role of ARS2 in effector versus memory T cell differentiation – a fate-tracking experiment observed approximately 3x higher ARS2 expression in effector-destined cells compared to their memory-destined counterparts (Kakaradov 2017). Does ARS2 therefore negatively regulate memory T cell formation while promoting effector differentiation? Furthermore, how do the different CD28 intracellular domains contribute to immunotherapy outcomes? The YMNM-PI3K motif was shown to be a primary target for PD-1-mediated dephosphorylation (Hui 2017, Kamphorst 2017), and this domain is required for glucose uptake immediately after activation (Frauwirth 2002), but we found this domain to be largely dispensable for anti-tumor immunity, and also found the PYAP-Grb2/Lck motif to be required for anti-PD-1 therapy. It stands to reason that both domains are involved in both non-redundant and overlapping functions; hopefully the sequencing datasets generated in this manuscript will serve as a resource to the immunology community as a whole to begin to answer some of these questions.
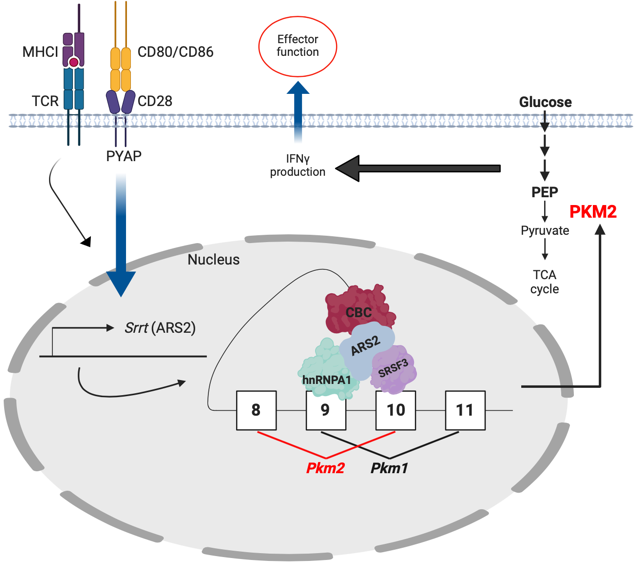
Figure 1: Proposed model of how ARS2 facilitates anti-tumor immunity. TCR binding antigen plus CD28 costimulation results in upregulation of ARS2 (Srrt). ARS2 then serves as a scaffold between the Cap-Binding Complex (CBC) and splicing factors to facilitate alternative splicing of Pkm to the Pkm2 isoform which, due to its lower rate constant than PKM1, promotes the buildup of upstream glycolytic intermediates, allowing for inflammatory cytokine production and anti-tumor effector responses.
Follow the Topic
-
Cellular & Molecular Immunology
A monthly journal from the Chinese Society of Immunology and the University of Science and Technology of China, covering both basic immunology research and clinical applications.

Ask the Editor - Immunology, Pathogenesis, Inflammation and Innate Immunity
Got a question for the editor about the complement system in health and disease? Ask it here!
Continue reading announcement


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in