Chemistry-informed recommender system to predict optimal molecular receptors in SERS nanosensors
Published in Chemistry, Materials, and Computational Sciences
The Spark: COVID-19 and the Trial-and-Error Problem
The story began during the early days of the COVID-19 pandemic, when we were developing a nanosensor to detect the virus. Like many in the field, we faced an overwhelming challenge: which molecular receptors should we use to capture the viral signature? The answer wasn’t clear, and the process of testing different recognition elements one by one was long, painstaking, and resource-intensive. I begin wondering: do we have to repeat this same trial-and-error process every time a new pathogen emerges? When the next disease “Disease X” comes, how can we respond faster? Is there a smarter way to use what we’ve already learned to tackle what we haven’t seen yet? Can we turn to data? Can we learn from previous sensor–analyte interactions to predict the best receptor candidates for new targets?
It sparked on day, can we we apply the same logic that powers recommender systems in our daily digital lives, like suggesting movies, song or instagram reels and use it to guide molecular recognition in nanosensors? What began as a frustrating bottleneck became the seed for a new approach: a data-driven, chemistry-informed recommender system for nanosensor design. Recommender systems have become integral to daily life. Whether suggesting the next movie to watch, product to buy, or article to read, these data-driven algorithms use past preferences to predict future behavior. While their success in consumer technology is well known, we began wondering whether a similar approach could be applied to molecular sensor design. Could a recommender system be used not to match people with products, but to pair chemical analytes with the right molecular receptors for sensing applications?
The Challenge of Receptor Selection in SERS
We explored this idea in the context of surface-enhanced Raman scattering (SERS), a technique that amplifies molecular vibrations near plasmonic metal surfaces. SERS offers high sensitivity but often struggles to distinguish between structurally similar molecules. One common solution is to functionalize the SERS substrate with molecular receptors, small chemical groups that selectively bind specific analytes, bringing them close to the metal surface for signal enhancement. The problem lies in selecting the right set of receptors. This is typically done through chemical intuition or trial and error, which becomes inefficient as the number of potential analytes and receptors grows.
Chemistry Is Predictable: A Case Study with Haloanisoles
We proposed a new approach: to use a data-driven, chemistry-informed recommender system to identify the best receptors for a given sensing task. Unlike human preferences, which are highly individual and context-dependent, molecular interactions are governed by clear rules of structure, polarity, and bonding. Because chemical reactivity is more predictable than human taste, we believed that a recommender system could be even more effective in this scientific domain.To test this concept, we focused on haloanisoles, a class of trace environmental contaminants that include compounds like 2,4,6-trichloroanisole and 2,4,6-tribromoanisole. These molecules are structurally very similar, differing only by the number and position of halogen atoms, yet they have distinct impacts, particularly in the context of food spoilage and odor contamination. Discriminating between them is difficult using standard SERS because their vibrational spectra are nearly identical. We hypothesized that by using a diverse set of molecular receptors and analyzing the resulting spectral variations through a recommender framework, we could significantly improve classification accuracy.
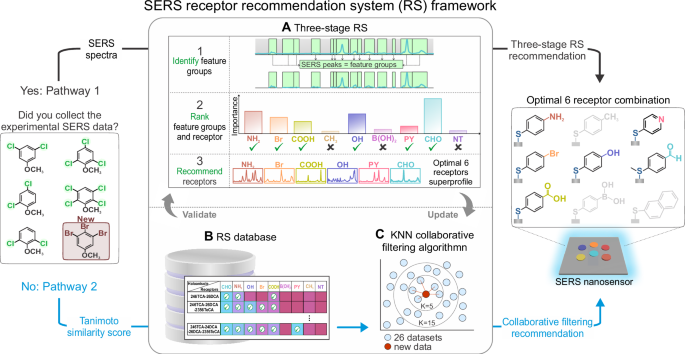
We began by constructing a library of nine small-molecule receptors, each with chemically distinct functional groups such as aldehyde, amino, hydroxyl, pyridine, and carboxylic acid. These groups were chosen to promote specific non-covalent interactions with the haloanisoles, including hydrogen bonding, dipole-dipole interactions, and halogen bonding. Each receptor was self-assembled onto the surface of silver nanocubes to create a set of functionalized SERS substrates. We then exposed each substrate to five different haloanisoles and collected over 1600 SERS spectra in total.The resulting spectral dataset captured subtle, analyte-dependent changes in key vibrational regions. For instance, we observed that the carbonyl stretch in the aldehyde receptor (CHO) shifted position depending on the analyte, consistent with changes in dipole-dipole interaction strength. These chemically meaningful shifts were validated using density functional theory (DFT) calculations and formed the basis of what we call a SERS superprofile: a composite, multi-receptor signal profile rich in discriminative information. But how many receptors are actually necessary? While adding more receptors increases the amount of data, it also introduces redundancy and can degrade machine learning performance through the so-called curse of dimensionality. We needed a principled way to identify which receptors contributed the most useful information and which could be excluded.
A Recommender System for Receptor Selection
To do this, we developed a three-stage machine learning pipeline inspired by recommender systems. In the first stage, we used spectral preprocessing and expert-guided filtering to identify chemically relevant feature groups from each receptor spectrum. These features were then evaluated using XGBoost, a gradient-boosted decision tree algorithm, to assess how much each feature contributed to accurate analyte classification. This allowed us to rank not just individual spectral features but also entire receptors based on their information value. In the final stage, we evaluated all 511 possible combinations of the nine receptors and identified which subset achieved the best classification accuracy. The optimal set included six receptors—CHO, NH2, OH, Br, COOH, and PY—and achieved 96.6 percent accuracy in classifying the five haloanisoles. Adding more receptors beyond this point actually reduced accuracy, confirming that an intelligent recommender strategy outperforms brute-force or intuition-based approaches.
Predicting Receptors for New and Untested Analytes
Having validated the system on known analytes, we wanted to push further: could the recommender predict the right receptors for new molecules that had not been part of the original training set? To enable this, we constructed a receptor–analyte matrix using classification data from 26 different haloanisole combinations and calculated pairwise Tanimoto similarity scores between molecules. These scores quantify how similar two molecules are based on their structure, and therefore how likely they are to interact similarly with a given receptor. Using a collaborative filtering model with k-nearest neighbors, we could then recommend receptors for a new analyte based solely on its structural similarity to known compounds. For example, the model correctly predicted the optimal six-receptor set for 2,4,6-tribromoanisole, a molecule not used in the original training. Experimental validation showed a classification accuracy of 95.7 percent, demonstrating that structural similarity is a reliable predictor of interaction behavior in this system.
Toward Smart, Data-driven Sensor Design
This chemistry-informed recommender system represents a shift in how we approach sensor design. By grounding our framework in well-understood chemical principles, we ensure that the machine learning models remain interpretable and generalizable. The receptors are not black-box features, but chemically defined entities with predictable behaviors. This transparency allows us to trust the system’s recommendations and understand the underlying reasons for their success. Moreover, the approach is scalable and broadly applicable to other recognition elements for various sensor modalities. While our proof-of-concept focused on haloanisoles in ethanol, the method is agnostic to analyte class and adaptable to more complex environments for multiplex sensing.
In conclusion, by combining the structured logic of chemistry with the predictive power of recommender systems, we have created a new paradigm for molecular sensor design. Unlike inherently unpredictable entertainment preferences, molecular interactions follow rules. That makes them ideal for data-driven systems that rely on consistency and repeatability. As recommender systems continue to shape our digital lives, they may also become essential tools in scientific discovery, helping us design smarter, faster, and more reliable nanosensors.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in