Computer simulations reveal tunneling pathways leading to hydrogen-bond rearrangement in the iodide-dihydrate complex
Published in Chemistry


This discovery came about accidentally as a result of staring at a computer screen watching molecules move on pico-second time scales under the influence of molecular interactions, following Newton’s laws of motion. 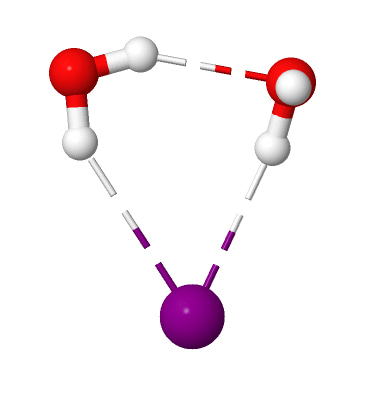 That’s something we do quite often as computational chemists. In this case, the particular molecular dynamics simulation we were looking at was that of an iodide ion with two water molecules (iodide-dihydrate) at 10 K temperature. At such low temperatures molecular systems tend to just vibrate about their lowest energy configuration since there isn’t enough energy for any large-scale motions. For the iodide-dihydrate complex, the minimum energy configuration is a cyclic arrangement with the two water molecules hydrogen-bonded (H-bonded) to the ion and to each other.
That’s something we do quite often as computational chemists. In this case, the particular molecular dynamics simulation we were looking at was that of an iodide ion with two water molecules (iodide-dihydrate) at 10 K temperature. At such low temperatures molecular systems tend to just vibrate about their lowest energy configuration since there isn’t enough energy for any large-scale motions. For the iodide-dihydrate complex, the minimum energy configuration is a cyclic arrangement with the two water molecules hydrogen-bonded (H-bonded) to the ion and to each other.
This simulation was one of several we ran at different temperatures to study the evolution of the iodide-dihydrate complex with increasing temperature. Previous studies have shown that at ~100 K the water-water H-bond breaks and the cluster transitions to a high energy dynamic configuration. Our goal was to provide molecular level insights into this process from our computer simulations.
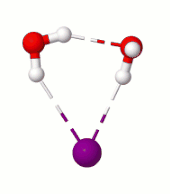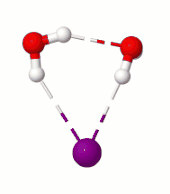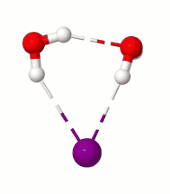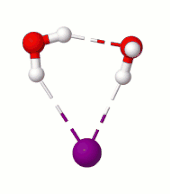
Surprisingly, in the simulation at 10 K we saw a rare rotational flip of the water molecule with one free OH bond and the other OH bond H-bonded to iodide. The rotational motion led to an “exchange” of the two OH bonds while breaking the iodide-water H-bond, which requires ~0.4 kcal/mol of potential energy. According to classical mechanics, this motion would not occur at 10 K (equivalent to ~0.02 kcal/mol). So, we suspected that what we are seeing has to be tunneling, a purely quantum mechanical phenomenon where due to the wave-like nature of the particles there is a finite probability for the system to cross the (potential energy) barrier even at low temperatures by “tunneling” through it instead of going over it.
However, the simulation alone is not enough as definitive proof of tunneling. We needed a more concrete approach. This led to the collaboration with Dr. Jeremy Richardson who had worked extensively on tunneling in molecular systems and developed a software to identify tunneling pathways and calculate tunneling splittings. Using his software we were able to identify, for the first time, not just one but three tunneling pathways in the iodide-dihydrate complex. In this study, we report the corresponding ground-state tunneling splitting pattern and tunneling timescales which provide quantitative evidence for the existence of the tunneling pathways. We also provide an in-depth analysis of nuclear quantum effects on the tunneling dynamics using isotopic substitution.
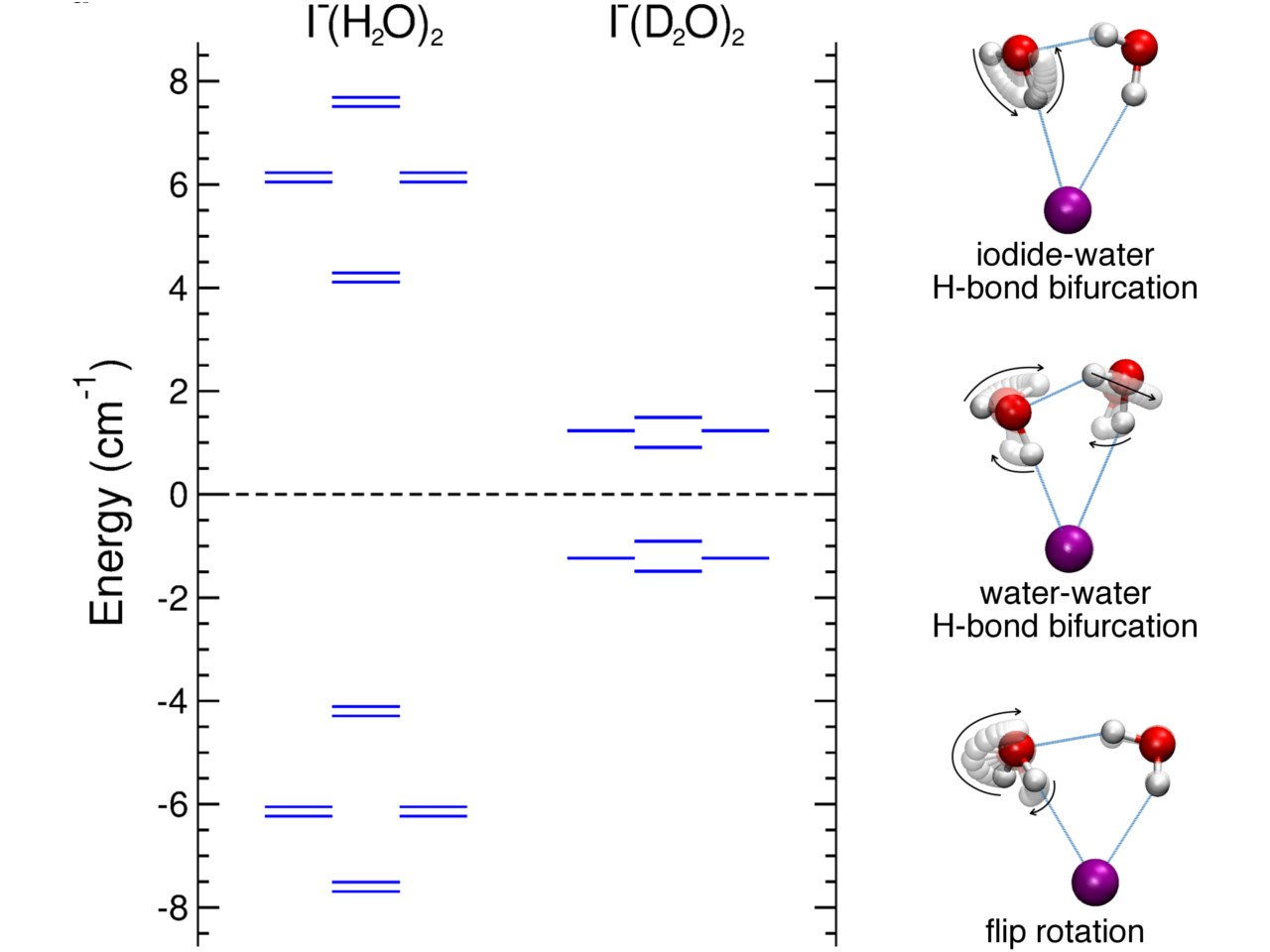
Similar tunneling pathways have been reported previously for the pure water trimer complex. Our results show that, in comparison to the pure water trimer, the presence of iodide ion significantly speeds up the large amplitude hydrogen bond rearrangement pathways as a result of weakening of the water-water H-bond.
Interestingly, this result is apparently in contrast with ultrafast vibrational spectroscopy measurements which suggest that the H-bond rearrangement dynamics within the first hydration shell of iodide ion is slowed down in bulk aqueous solution compared to that in pure water. A plausible explanation for this difference is that the free OH bond present in the iodide-dihydrate complex essentially initiates the overall H-bond rearrangement motions and enables faster reorientations of the four OH bonds. Along these lines, one could imagine similar factors coming into play at the interfacial regions of salt solutions where the hydration structure can be under-coordinated with at least one dangling free OH bond.
We believe that this study provides an example of the rapidly growing potential of computer simulations in chemistry. In the case of the iodide-dihydrate complex, we show that state-of-the-art quantum simulations not only reproduce available experimental data but also provide a rigorous interpretation of the underlying molecular mechanisms and allow us to predict tunneling splittings that cannot be observed using existing experimental techniques due to the lack of necessary resolution.
By Pushp Bajaj
Follow the Topic
-
Nature Chemistry

A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in