Connecting the Dots: Epigenetic Abnormality and Chromosomal Instability in Cancer
Published in Cell & Molecular Biology
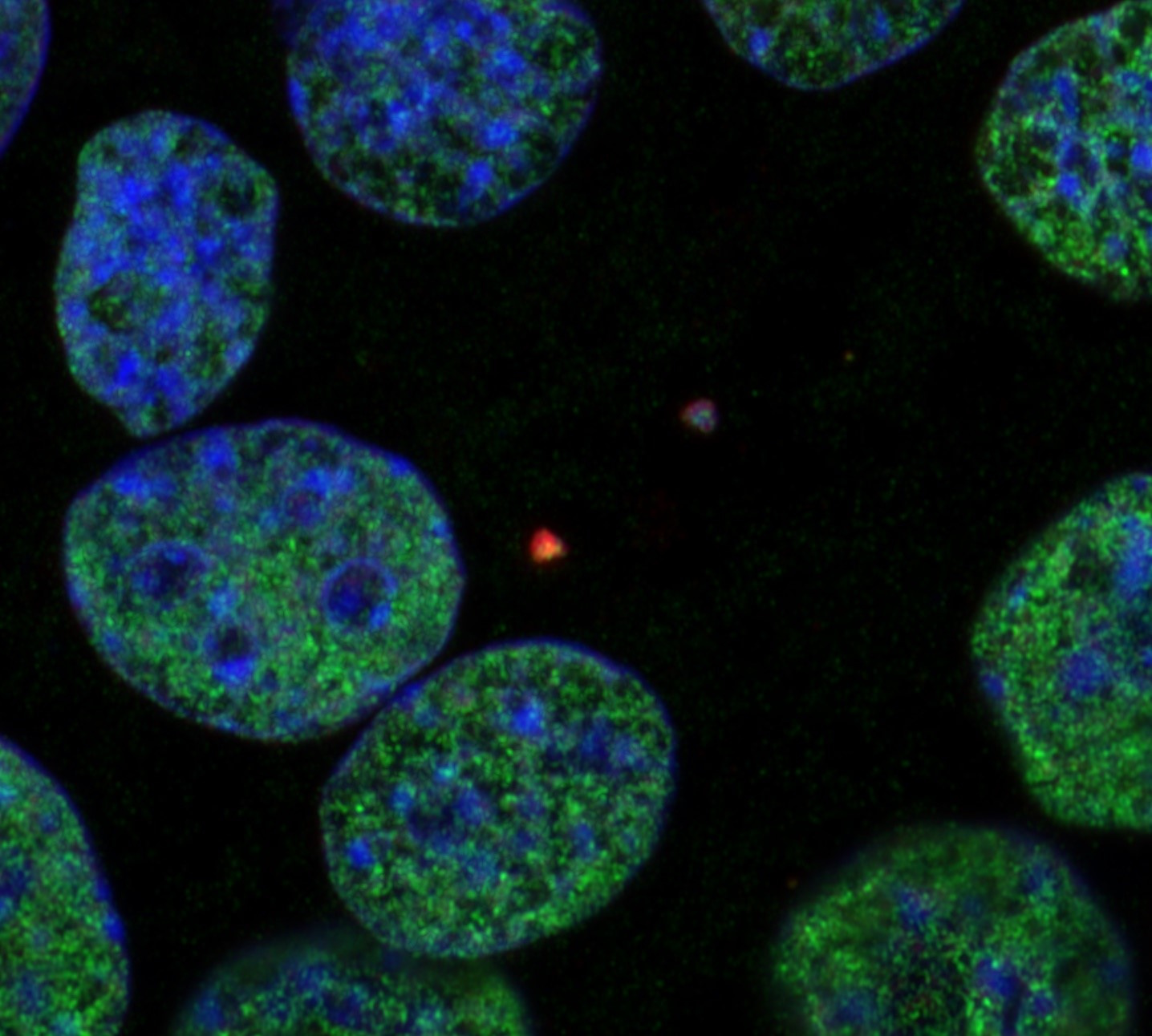
Charles Darwin’s theory of natural selection is one of the cornerstones of modern biology. The concept asserts that organisms boasting diverse phenotypes have a higher chance of surviving and reproducing when exposed to environmental pressure, thereby creating a way for organisms to adapt and change. Intriguingly, cancer cells exploit this principle by leveraging mechanisms akin to natural selection to thrive, adapt, and resist treatment. A crucial aspect of this process is the generation of genomic diversity, often achieved through the mechanism known as chromosomal instability (CIN).
CIN is one of the hallmarks of cancer. It is characterized by ongoing errors in chromosome segregation during cell division. These missegregated chromosomes often end up recruiting their own nuclear envelope and forming a distinct structure in the cell dubbed micronuclei. This phenomenon generates genomic copy number heterogeneity, as one daughter cell might get a whole or fragments of a missegregated chromosome, and the other daughter cell loses them. In chromosomally unstable cells, this event happens continuously, creating a diverse array of cells with different chromosomal copy numbers. In addition, chromosomes in micronuclei can undergo severe DNA damage, which can result in genomic rearrangement.
On a molecular scale, the smallest unit of a chromosome is a nucleosome, a structure consisting of DNA wrapped around histone proteins. The organization of nucleosomes in a higher-order structure allows for the compaction of the genetic material in the nucleus. Beyond DNA organization, the packaging histone proteins undergo extensive post-translational modification (PTM) that dynamically regulates gene expression as well as DNA damage repair and replication. In cancer, histone PTMs are frequently dysregulated. Indeed, efforts to correct this mechanism in cancer have led to several FDA-approved drugs such as vorinostat and tazemetostat.
The impetus of the research was our initial question of whether epigenetic changes can be introduced in CIN, thereby creating “epigenetic heterogeneity” on top of “genomic heterogeneity” and essentially mechanistically linking CIN and epigenetic dysregulation. From that point on, we embarked on an intriguing journey, with new revelations in every corner as we marched towards marrying two fields that previously had only loose ties connecting them.
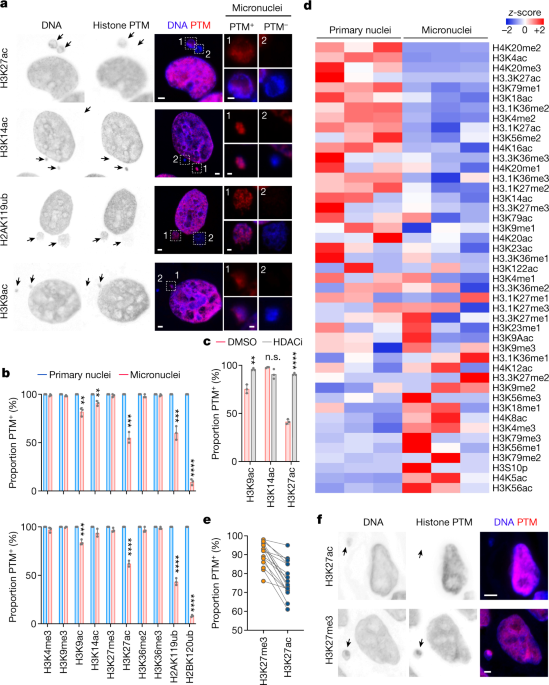
We began our efforts by answering a simple question, whether histone PTMs in micronuclei are different compared to the ones existing in the primary nuclei. Using immunofluorescence staining for several key histone PTMs, we compared their abundance. We were very excited to observe that not only were they different, but the types of histone PTMs that had changed were strikingly consistent between the tested cell lines – including human and mouse as well as cancer and non-transformed cells. This was later orthogonally confirmed using a quantitative unbiased mass spectrometry profiling of histone PTMs in isolated micronuclei and primary nuclei. Moreover, the changes in histone PTMs persisted even after the chromatin from micronuclei was reincorporated into primary nuclei in subsequent cell cycles. Importantly, these changes were also observed in patient tumor samples, underscoring their clinical relevance.
We next wondered what might be the mechanism that governs these differences. Our attention turned to the lifecycle of the micronucleus. During interphase, micronuclei often undergo envelope rupture which exposes their contents to the cytosol. We hypothesized that this event could have an impact on histone PTMs, given their abnormal exposure to the cytoplasmic compartment. It seemed to indeed be the case for most of the histone H3 acetylation changes that we observed (K9ac, K14ac, and K27ac) alongside histone H2A K119 ubiquitination, where we saw loss of these PTMs only in ruptured micronuclei. However, we observed that other changes were unaffected by the rupture, for example, histone H3 methylation gains and H2B K120 ubiquitination loss. These could be explained by the histone PTMs’ persistence (or absence) within lagging chromosomes during mitosis, prior to them becoming a micronucleus, when we observed them using immunofluorescence.
Since histone PTMs modulate chromatin accessibility, our attention naturally shifted towards deciphering how dysregulation of histone PTMs in micronuclei might be translated to changes in chromatin accessibility. Employing a suite of orthogonal methods, we observed that chromatin was more compacted in micronuclei, which was in line with the histone PTM changes trend we observed. However, diving deeper into the data, we found that there was a positional bias in micronuclei chromatin accessibility. Promoter and transcription start site regions seemed to be more accessible while gene bodies and intergenic regions were less accessible. We managed to attribute some of this positional bias to a particular histone PTM: histone H3K4 trimethylation (H3K4me3), a histone PTM that correlates with open chromatin region and is often associated with highly transcribed genes. When mapped to the genome, this histone PTM’s location in micronuclei mostly mirrored the open regions that were initially found in the micronuclei. This finding served as an important link between changes in histone PTM and chromatin function.
Finally, and perhaps the most important question, was to understand the role histone PTM changes play in regulating chromatin accessibility in chromosomally unstable cancer cells. To answer this, we induced missegregation and micronucleation of specific chromosomes and analyzed the epigenetic changes after multiple cellular generations. Gratifyingly, the epigenetic dysregulation that we observed happened exclusively in the micronucleated chromosomes with varying abnormalities in individual cells. Remarkably, we found that this phenomenon lasted for at least 30 days. These results imply that chromosomal instability acts as a driver for heritable, lasting epigenetic dysregulation, on top of genomic alterations and that cancer cells possess a dual strategy to diversify both their genome AND epigenome to further increase their odds to thrive, adapt, and resist treatment.
The work highlights the critical advantages of multidisciplinary cellular & molecular biology research, particularly in the context of our understanding of cancer. This was a joint collaboration between investigators whose expertise spanned cancer biology and chemical-biology, bringing forth their unique expertise to address the important question of the link between epigenetics and CIN. We are all extremely grateful to also be able to have the chance to collaborate with various experts from multiple institutions, without which, we would not be able to employ important cutting-edge techniques and analyze the critical data that we produced. It is important to note that in parallel to our work, Papathanasiou et al. showed transcriptional heterogeneity from micronuclei and cytoplasmic chromatin. These two complementary articles reveal critical mechanisms of chromatin structure, histone PTMs, and transcriptional changes in CIN and open the door to many follow-up works deconvoluting the mechanisms of these phenomena. Finally, these discoveries open the door to exploring the role epigenetics plays in regulating chromosomal abnormalities and furthermore, the effect and use of epigenetic drugs to target CIN in cancer.

Follow the Topic
-
Nature
A weekly international journal publishing the finest peer-reviewed research in all fields of science and technology on the basis of its originality, importance, interdisciplinary interest, timeliness, accessibility, elegance and surprising conclusions.

Related Collections
With Collections, you can get published faster and increase your visibility.
Carbon Dioxide Removal
Publishing Model: Hybrid
Deadline: Jan 16, 2027

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
awosome