CryoREAD: a fully automated DNA–RNA structure modeling tool for cryo-EM maps
Published in Protocols & Methods

Cryogenic Electron Microscopy (cryo-EM) revolutionized structural biology with its superior ability to determine macromolecules. Consequently, there are many structure modeling software developed to aid 3D structure model building from a cryo-EM map; however, most of these are for protein structure determination. Compared to proteins, nucleic acids are flexible and typically exist in complex with proteins, thus presenting substantial challenges for accurate modeling in a cryo-EM map of a medium resolution of about 5 Å. DNA/RNA is often modeled manually with interactive software. An automated, efficient, and accurate modeling tool for nucleic acid structures would greatly assist the modeling process. To address this need, CryoREAD was developed for fully automated DNA/RNA structure modeling from cryo-EM maps.
Kihara Lab is an interdisciplinary research group affiliated with both in biology and computer science (CS) departments. Our lab physically locates in the structural biology building, and we have observed numerous successful structural biology projects through cryo-EM in the last decade. We also worked on the cryo-EM related software development for more than six years. We have developed several tools for structure modeling (MAINMAST, MAINMAST-Seg, DeepMAINMAST), structure detection (Emap2sec, Emap2sec+), structure evaluation (DAQ), structure refinement (DAQ-Refine) and map alignment (VESPER). They are all freely available on our EMSuites and EM-Server.
![]()
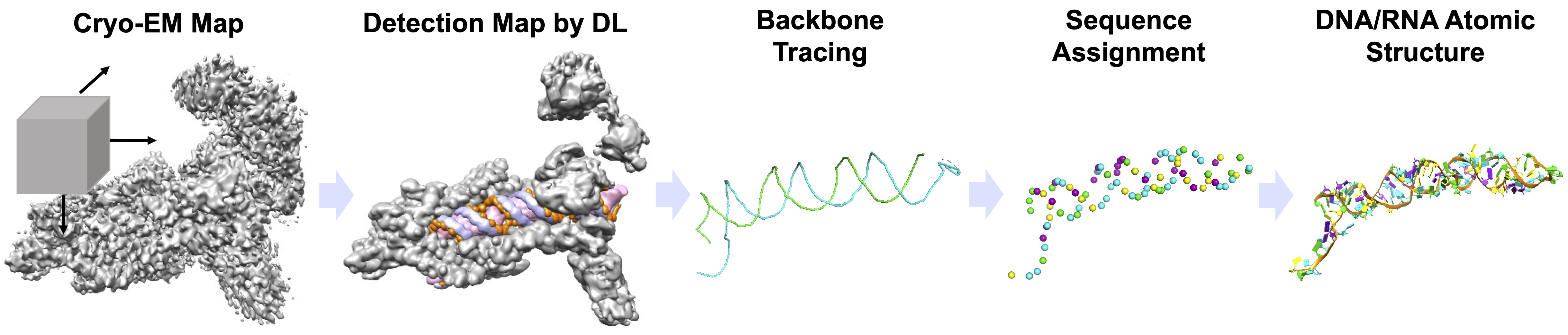
CryoREAD uses deep learning to identify key structural information in a cryo-EM map. First, deep neural networks accurately identify the possible locations of sugars, phosphates, bases and base types in the map by capturing their characteristic local density patterns. The identified sugar positions are then connected to form the backbone structure of nucleic acids. Subsequently, the nucleic acid sequence is mapped along the backbone by considering the identified base types along the traced backbone. Finally, a full atomic structure, including nucleotide bases, is constructed. The modeling process does not require human intervention such as parameter tuning or manual modeling.
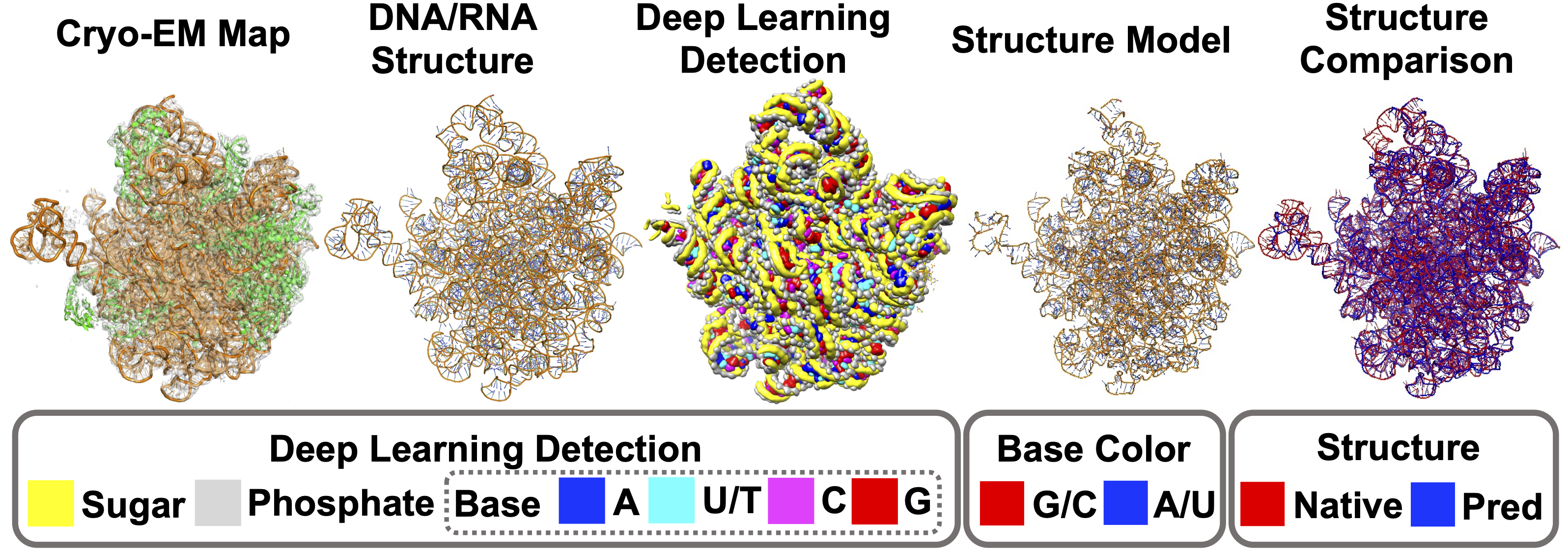
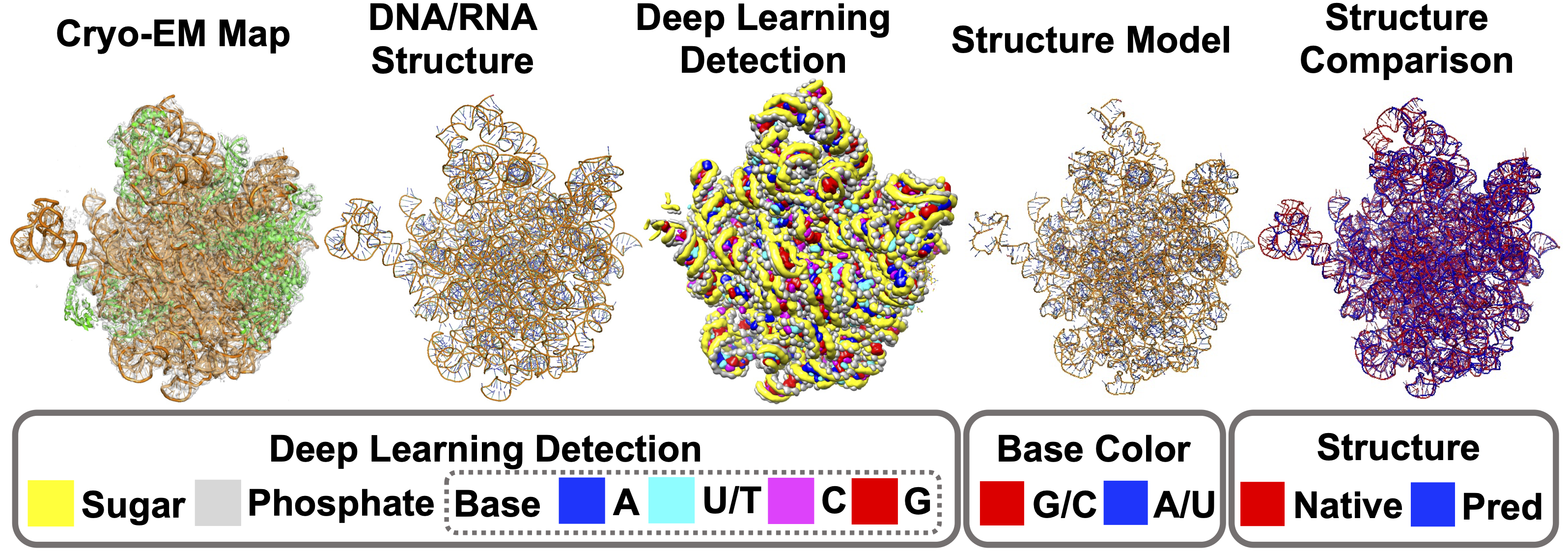
We first tested CryoREAD on a dataset with 11 DNA entries, 55 RNA entries and 2 DNA–RNA mixed entries. Backbone accuracy and sequence accuracy were 85.7% and 52.5%, respectively. We further evaluated CryoREAD on 61 maps including RNA entries from SARS-CoV-2 and observed similar performance. Particularly, CryoREAD can model big protein-RNA complex structure very well. For example, the figure below is a bacterial pre-50S ribosomal precursor complexed with ribosomal silencing factor RsfS and GTPase ObgE/CgtA. This large complex includes 3,702 amino acids and 3,016 nucleotides, and the map (EMD-12217) is determined at a resolution of 2.4 Å. Despite the large size, CryoREAD was able to separate RNAs from proteins, resulting in a full atom model with a high backbone recall of 0.910. As shown in the "Deep Learning Detection" panel, the detected sugars and phosphates by deep learning traced the backbone structures of RNAs well, which laid a solid foundation for accurate RNA structure modeling.
To help structural biologists to build DNA/RNA structures from cryo-EM maps, the webserver is available at https://em.kiharalab.org/algorithm/CryoREAD, where users can simply upload the map and obtain the structures without installment. Full source code is available in Github: https://github.com/kiharalab/CryoREAD. Users can also access Google Colab Notebook webserver at https://bit.ly/CryoREAD. A detailed tutorial for CryoREAD is available at https://kiharalab.org/emsuites/cryoread.php. If you have any questions or possible ideas to further improve CryoREAD, please contact Prof. Daisuke Kihara (dkihara@purdue.edu).
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Oct 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in