DAQ: A deep-learning based quality assessment tool for protein models from cryo-EM maps
Published in Protocols & Methods

Cryogenic Electron Microscopy (cryo-EM) revolutionized structural biology with its superior ability to determine macromolecules. Meanwhile, with the advance of cryo-EM, its resolutions and reconstructions are also quickly improving. In the Election Microscopy Data Bank (EMDB), a public database for cryo-EM, 58% of the maps have near atomic resolution (>=4 Å) compared to only 31% in 2017.
Though the overall improved resolution resulted more and more accurate structures, it’s still often challenging to have correct amino acid assignments The “resolution revolution” of cryo-EM has opened the door of structural analysis to many less experienced users who are attempting to build atomic models into maps of moderate resolution and widely varying local resolution. Therefore, it’s in pressing need for rigorous validation of the resulting atomic model if one wants to produce the most accurate model possible from the data in hand. Therefore, we present our new approach, Deep-learning-based Amino acid-wise model Quality (DAQ) score, for cryo-EM protein model validation.
KiharaLab is an interdisciplinary research group affiliated both in biology and computer science (CS) departments. Our lab physically locates in the structural biology building, and we have observed numerous successful structural biology projects through cryo-EM in the last decade. We also worked on cryo-EM related software development with Emap2sec, Emap2sec+, MAINMAST, MAINMAST-SEG, EM-GAN, VESPER. Their introductions and possible applications are included in our em-suite website. That accumulated rich experience for us on applying new algorithms, particularly deep learning, to process cryo-EM maps. Thus, it’s natural for us to apply deep learning for structure quality assessment of protein models from cryo-EM maps.
![]()
Deep-learning-based Amino acid-wise model Quality (DAQ) score computes the likelihood that the local density corresponds to different amino acids, atoms, and secondary structures, estimated via deep-learning, and assesses how well the amino acid assignment in the atomic protein structure model is consistent with that likelihood. We used deep learning because our previous success in Emap2sec, Emap2sec+ have suggested that underlined molecular structure in an EM map can be detected by deep learning from map density. Our ongoing research also suggests that such information is useful to guide structure modeling. Therefore, we trained a deep convolutional neural network that can predict protein secondary structure, amino acid type and atom types at the same time via multi-task training. Then the local predicted features by deep learning are compared with amino acids in the structure built from the EM map to computer DAQ scores. DAQ score can indicate if an amino acid residue assigned to a local density is likely to be incorrect, even in cases where the protein sequence is misaligned along an otherwise correct main-chain trace. Our results suggest that incorrect amino acid assignment can happen even when the residue has reasonably high local density cross-correlation and appropriate stereochemical geometry. For such cases, previous methods based on map-model correlation or geometry model-coordinate evaluation can’t recognize while DAQ can detect them successfully.
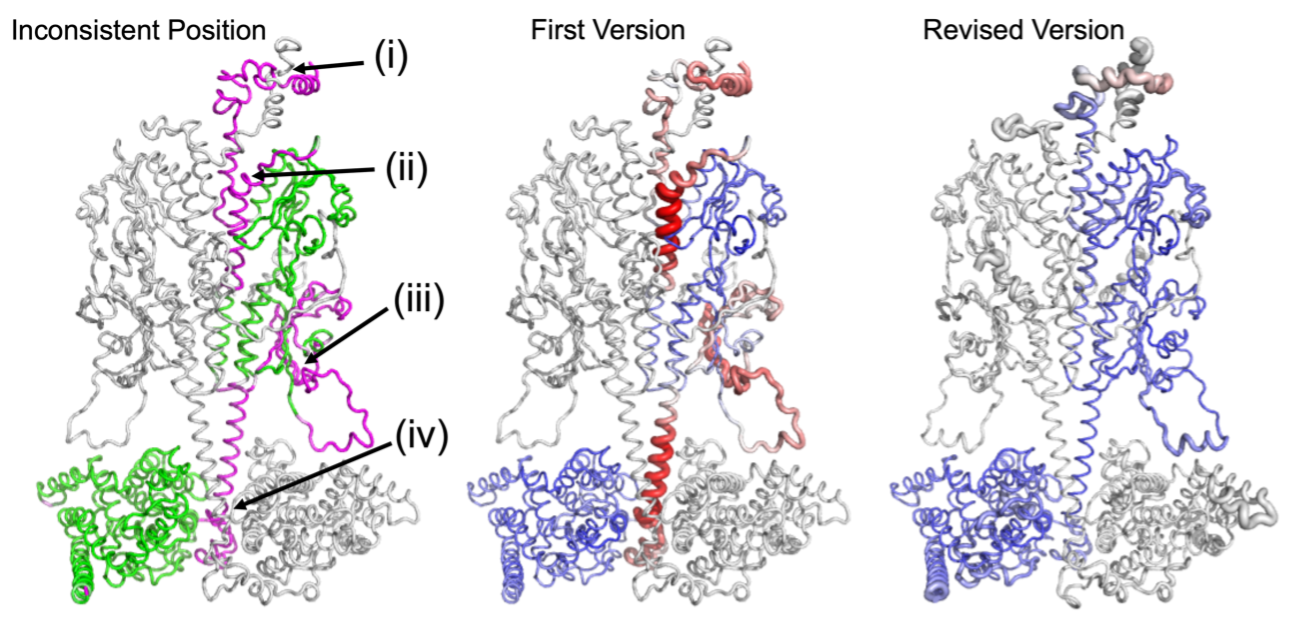
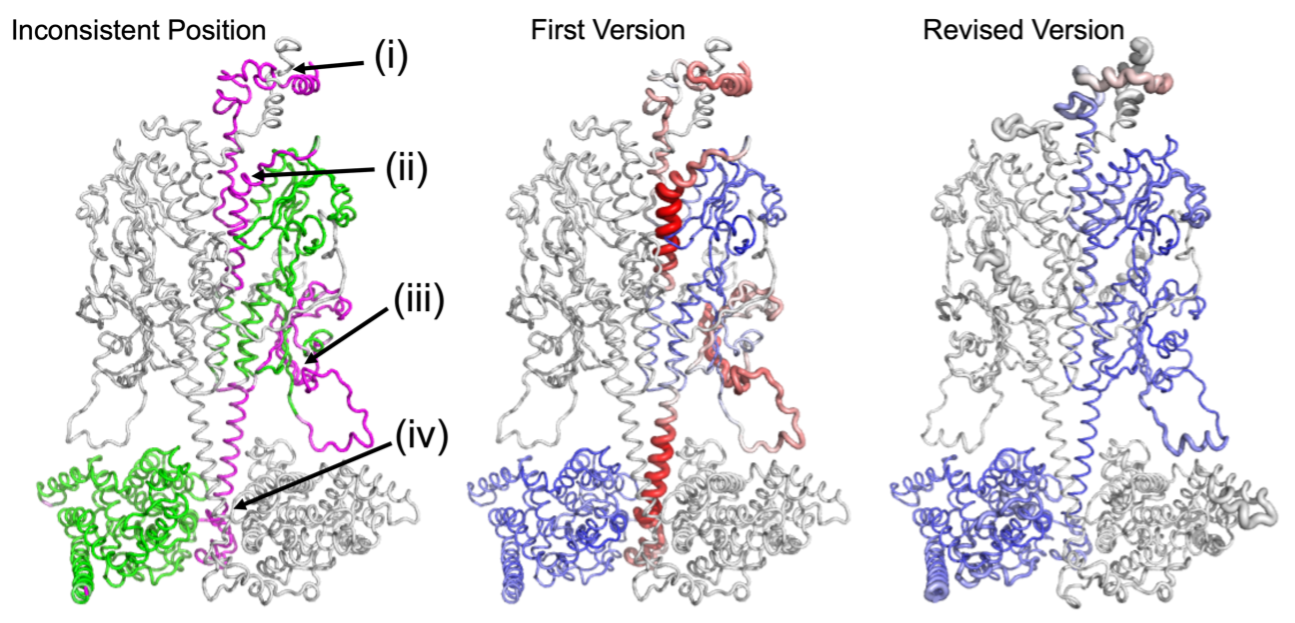
To verify the effectiveness and reliability of DAQ, we applied DAQ on several different settings. Because of some possible structure errors, sometimes authors will upload more than 1 version structure for the same cryo-EM map. For such structures, we found that in most cases, the later version of the deposited structure has a better DAQ score than the corresponding first version of the models. That indicates the revised models were typically improved and DAQ is reliable for local quality assessment. We further tested DAQ on 399 pairs of PDB entries of protein structures of high sequence identity built from cryo-EM maps in which the models differ by more than 1 Å RMSD from each other. We found that most of the pairs have a large difference in their DAQ scores, strongly implying that one (or both) of the models may contain serious errors. Moreover, for 4,485 PDB models at better than 5 Å resolution, we observed 89 PDB-chain models (2.0%) have possible misassignments of more than 10% of the residues.
To help structural biologists to improve the structures from cryo-EM maps, we have full released our code in https://github.com/kiharalab/DAQ and we also provided an online platform https://bit.ly/daq-score for online structure quality assessment. If you have any questions or possible ideas to further improve DAQ, please make contact with Prof. Kihara (dkihara@purdue.edu).
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Oct 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in