Deciphering the mechanism of the Ni-photocatalyzed C‒O cross-coupling reaction using a tridentate pyridinophane ligand
Published in Chemistry
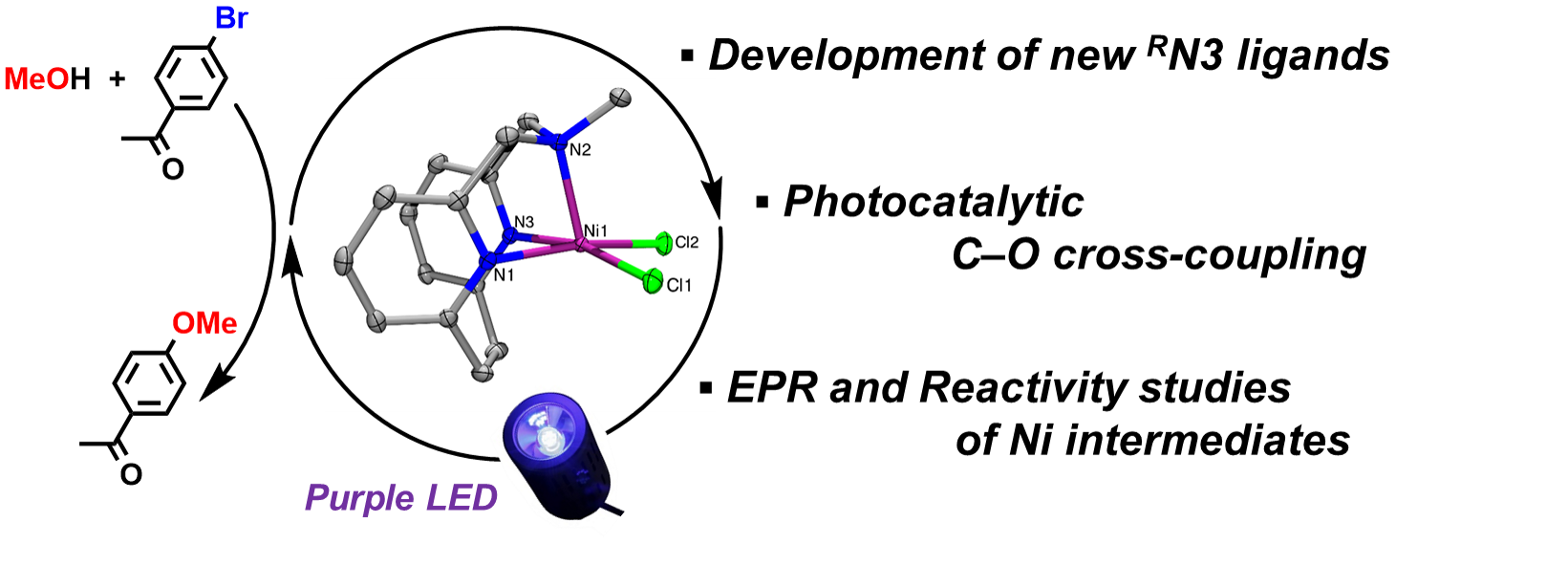
Over the past several decades, visible light-mediated photoredox catalysis employing Ni complexes has made vital contributions in the field of synthetic organic chemistry, specifically for C-C, C-N, C-O cross-coupling reactions[1-3]. In many research reports conducted in the realm of organic chemistry, developing new methodologies and reaction condition optimization are often done by trial-and-error, rather than based on a thorough understanding of the underlying reaction mechanism[4]. This might be because understanding of the underlying chemistry requires a great deal of contributions from the inorganic and organometallic chemistry fields (beyond the scope of the research interests in synthetic organic chemistry), such as the synthesis and characterization of related metal complexes, and study of their photochemistry and photophysics (Fig. 1).
Although a lot of effort to understand the mechanism of photocatalytic cross-coupling reactions has been made by both the organic and inorganic chemistry community, there are still several unanswered questions. For example, in the redox Ni catalytic cycle, the oxidation states of the involved Ni intermediates are elusive. In some cases, the involvement of paramagnetic NiI and NiIII species has been implicated, yet such intermediates have not been thoroughly investigated, and the key catalytic steps of oxidative addition, transmetalation, and reductive elimination have rarely been observed directly[2,3]. As such, our group, a synthetic inorganic and organometallic chemistry group, decided to develop a new ligand framewotk to stabilize various Ni species and use a range of inorganic spectroscopic and electrochemical techniques to decipher the mechanism of the Ni-photocatalyzed C–O cross-coupling reaction.
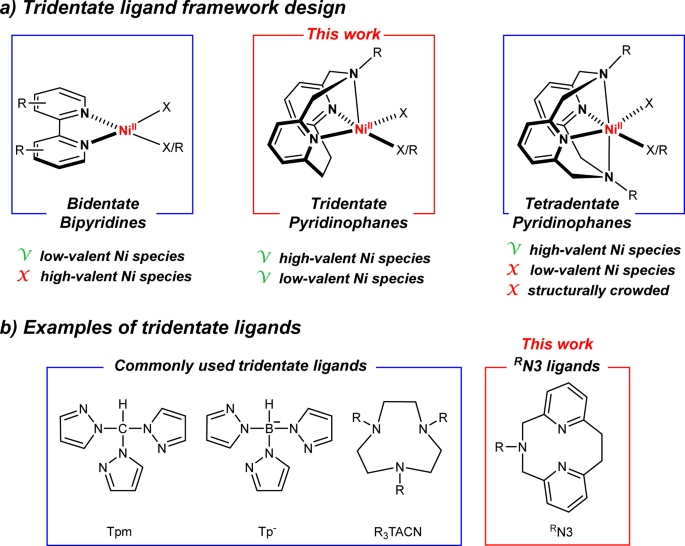
Our group has extensively employed tetradentate azamacrocycle N,N’-dialkyl-2,11-diaza[3.3](2,6)pyridinophanes (RN4) ligands (Fig. 2) to isolate and characterize NiIII species capable of C–C[5] and C–heteroatom[6] bond formation reactions, and to probe the involvement of high-valent Ni species in these reactions. Since the most ubiquitous ligands in Ni cross-coupling and photocatalytic reactions are the bidentate bipyridine ligands[1], the RN4 ligands were thought to be less effective ligands for catalytic applications due to the crowded coordination environment around the Ni center, which is also expected to disfavor the formation of NiI species. By contrast, a bidentate ligand structure is not suitable for stabilizing high-valent Ni species, thus hampering the investigation of such Ni intermediates[7]. Considering these aspects, we sought to identify an optimal ligand framework positioned in between bipyridine and RN4 ligands in terms of denticity, molecular structure, and functionality, and thus the pyridinophane tridentate RN3 ligands were targeted (Fig. 2). These RN3 ligands are structurally analogous to the RN4 ligands, possessing a rigid aromatic pyridinophane framework and containing only one flexible chelating arm that allows for either a κ2 or κ3 coordination.
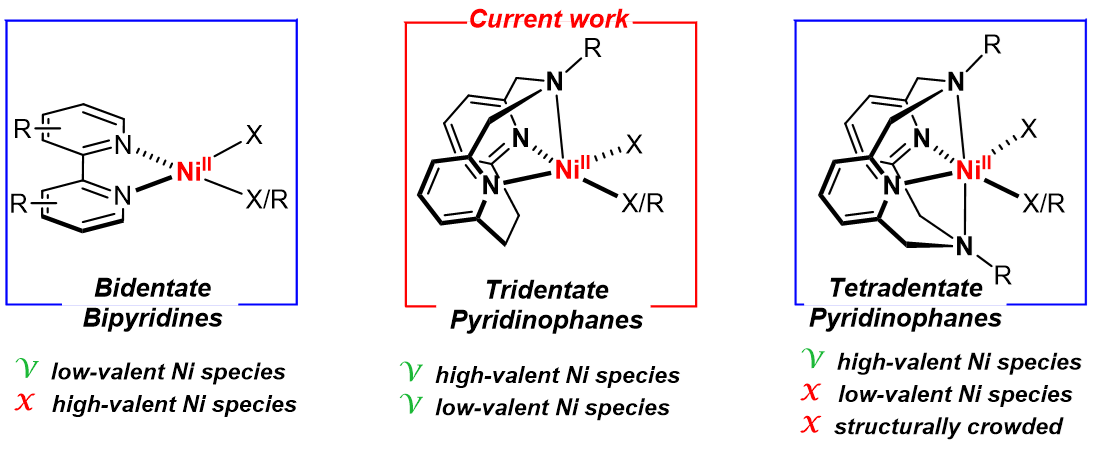
Figure 2. Design strategy used for the development of the tridentate ligand framework.
After considering different synthetic strategies to generate RN3 tridentate ligands, we have been successful in employing an N-extrusion reaction originally reported by Overberger[8], to successfully synthesize the unprecedented tridentate pyridinophane RN3 ligands (MeN3 and iPrN3 in this work). The derived (RN3)NiCl2 complexes (Fig. 3: structure of (MeN3)NiCl2 is shown) exhibit catalytic activity in C–O cross-coupling without any additional photocatalyst under purple LED irradiation (λmax = 390 nm), notably at higher substrate concentrations (1 M) and low catalyst loadings (0.2 mol%).
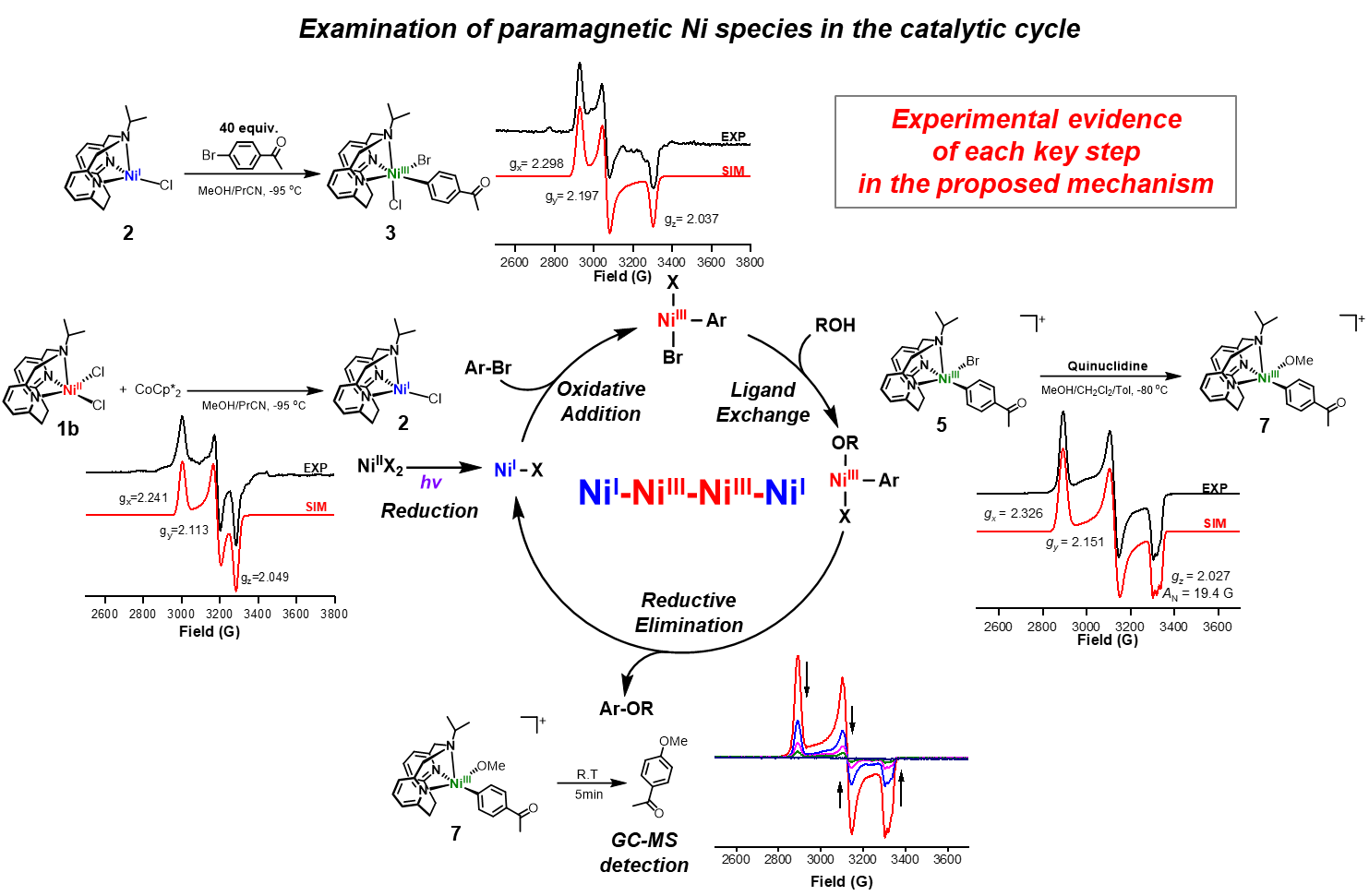
Since our Ni catalysts have accessible NiII/I redox potentials (as examined by cyclic voltammetry), we aimed to probe the reduction of NiII to NiI and the subsequent oxidative addition of an aryl halide to generate a NiIII intermediate by using X-band electron paramagnetic resonance (EPR) spectroscopy (Fig. 4). Upon the reduction of 1b by 1 equiv decamethylcobaltocene (CoCp*2) at –95 °C, a NiI species 2 was formed that exhibits a rhombic EPR spectrum, and suggesting the presence of a metal-based radical. The detection of an EPR-active mononuclear NiI species is important since such an intermediate is essential for an efficient oxidative addition step. Moreover, when 4-bromoacetophenone was added to 2, a rhombic EPR signal of the proposed transient 6-coordinate (iPrN3)NiIII(PhAc)BrCl (3) complex was observed and thus strongly supporting a direct conversion from NiI (2) to NiIII (3). While the exact structure of this species 3 is not unambiguously known, it is important to note that, to the best of our knowledge, the direct observation of the oxidative addition of an aryl halide to a NiI species to generate a NiIII species has not been reported to date.
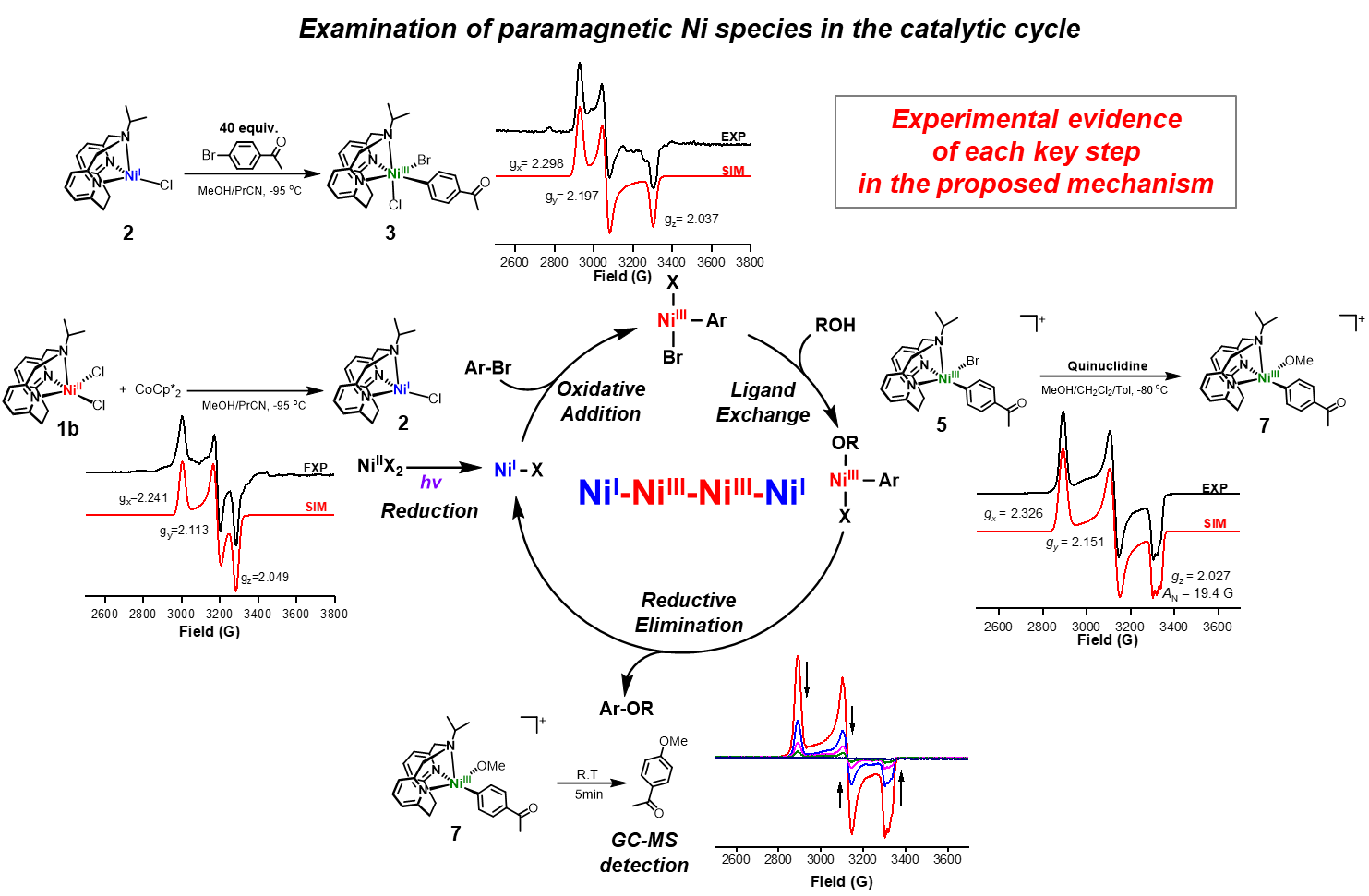
Figure 4. EPR studies of the odd-electron Ni species proposed in the catalytic cycle.
To further probe the role of various NiIII species in the C–O coupling reaction, we independently synthesized the NiIII complex [(iPrN3)NiIII(PhAc)(Br)]+ (5). The appreciable stability of 5, which is attributed to the presence of a tridentate ligand, allowed us to investigate its reactivity. First of all, we probed the ligand exchange step with an alkoxide. The addition at –80 °C of quinuclidine in MeOH to 5 generates a species assigned as the NiIII-aryl alkoxide complex [(iPrN3)NiIII(PhAc)(OMe)]+ (7). Importantly, to the best of our knowledge this is the first experimental observation of a ligand exchange reaction (either by a halide or alkoxide) occurring at a NiIII metal center, which is typically proposed as a key step in Ni-mediated C–C and C–heteroatom bond formation reactions[9,10]. Upon warming up to room temperature, species 7 undergoes rapid C–O bond-forming reductive elimination, affording 4-methoxyacetophenone and consistent with a favorable reductive elimination from a NiIII species[11,12].
Lastly, the same C–O coupling reaction was also probed under common dual Ni/photoredox catalytic conditions using various Ir or Ru photocatalysts and blue LED light (λmax = 456 nm), for a comparison of the redox potentials of the Ni complexes and various photocatalysts. This photocatalyst screening experiment revealed that an oxidative quenching pathway is proposed to be operative, in which the initial step is the single-electron reduction of NiII by the excited photocatalyst, and NiI and NiIII oxidation states are involved as catalytic intermediates. This observation is also consistent with the EPR spectroscopic studies supporting the NiI - NiIII catalytic cycle.
To conclude, we show that employing the newly developed tridentate RN3 ligands allowed us to directly probe the key steps of the C–O cross-coupling photocatalytic cycle involving paramagnetic Ni species: the oxidative addition of an aryl halide to a NiI species to generate a NiIII species, the ligand exchange/transmetalation step at a NiIII center, and the C–O bond-forming reductive elimination from a NiIII species. Overall, our work suggests that the RN3 ligands can lead to the development of new Ni catalysts, and are also a practical platform for detailed mechanistic studies of related Ni-catalyzed reactions. We hope that our work provides crucial insight into the reaction design and search for new chemical transformations in the burgeoning field of photoredox catalysis, and thus can impact both the organic and inorganic chemistry community.
This research was led by Prof. Liviu M. Mirica at the University of Illinois at Urbana-Champaign. More details on this work can be found in our manuscript “Deciphering the mechanism of the Ni-photocatalyzed C‒O cross-coupling reaction using a tridentate pyridinophane ligand” published in Nature communications. Link: https://doi.org/10.1038/s41467-022-28948-8
References
- Twilton, J. et al. The merger of transition metal and photocatalysis. Nat.ure Rev.iews Chem.istry 1, 0052 (2017).
- Shields, B. J., Kudisch, B., Scholes, G. D. & Doyle, A. G. Long-Lived Charge-Transfer States of Nickel(II) Aryl Halide Complexes Facilitate Bimolecular Photoinduced Electron Transfer. J. Am. Chem. Soc. 140, 3035-3039 (2018).
- Sun, R. et al. Elucidation of a Redox-Mediated Reaction Cycle for Nickel-Catalyzed Cross Coupling. J. Am. Chem. Soc. 141, 89-93 (2019).
- Arias-Rotondo, D. M. & McCusker, J. K. The photophysics of photoredox catalysis: a roadmap for catalyst design. Chem. Soc. Rev. 45, 5803-5820 (2016).
- Zheng, B. et al. Organometallic Nickel(III) Complexes Relevant to Cross-Coupling and Carbon–Heteroatom Bond Formation Reactions. J. Am. Chem. Soc. 136, 6499-6504 (2014).
- Zhou, W., Zheng, S. A., Schultz, J. W., Rath, N. P. & Mirica, L. M. Aromatic Cyanoalkylation through Double C-H Activation Mediated by Ni(III). J. Am. Chem. Soc. 138, 5777-5780 (2016).
- Camasso, N. M. & Sanford, M. S. Design, synthesis, and carbon-heteroatom coupling reactions of organometallic nickel(IV) complexes. Science 347, 1218-1220 (2015).
- Overberger, C. G., Lombardino, J. G. & Hiskey, R. G. Novel Reductions of N-Nitrosodibenzylamines—A New Reaction. J. Am. Chem. Soc. 80, 3009-3012 (1958).
- Shields, B. J. & Doyle, A. G. Direct C(sp3)–H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc. 138, 12719-12722 (2016).
- Sheppard, T. D. Metal-catalysed halogen exchange reactions of aryl halides. Organic & Biomol.ecular Chem.istry 7, 1043-1052 (2009).
- Matsunaga, P. T., Hillhouse, G. L. & Rheingold, A. L. Oxygen-atom transfer from nitrous oxide to a nickel metallacycle - synthesis, structure, and reactions of (2,2'-bipyridine)Ni(OCH2CH2CH2CH2). J. Am. Chem. Soc. 115, 2075-2077 (1993).
- Han, R. Y. & Hillhouse, G. L. Carbon-oxygen reductive-elimination from nickel(II) oxametallacycles and factors that control formation of ether, aldehyde, alcohol, or ester products. J. Am. Chem. Soc. 119, 8135-8136 (1997).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in