Domain-PFP allows protein function prediction using function-aware domain embedding representations.
Published in Protocols & Methods


Proteins act as the building blocks of life and govern the diverse activities in living organisms. From catalyzing biochemical reactions and transmitting signals within the body to providing structural support and transporting nutrients, proteins regulate biological processes and are the key players in the intricate orchestra of life (Fig. 1a). As a result, predicting protein function has been a longstanding problem in bioinformatics, given its plethora of downstream applications involving drug discovery and the understanding of disease and organic mechanisms.

Domains are the structural and functional units of proteins (Fig. 1b). Therefore, it is motivating to infer the functions a protein performs from the set of domains it comprises. Ongoing projects such as InterPro2GO, Pfam2GO, assign probable functions to the different domains through experimental validation. However, this approach is yet to be widely adopted for practical usage, primarily due to the lack of sufficient coverage, i.e., only a small subset of the domain-function relationships has been identified. As a result, machine learning-based methods have been developed to infer protein functions from the domains present within them.
The existing domain-based function prediction methods take the set of domains as input and aim to predict the set of functions in the form of Gene Ontology (GO) terms. However, both of these sets are quite large; for example, the InterPro database has approximately 14K domains, and there are more than 26K Gene Ontology terms. The high dimensionality of both the input and output space, coupled with insufficient functionally annotated proteins, hinders deep learning models from being sufficiently complex. Consequently, this results in high degrees of compression-expansion ratios within the network.
In order to avoid such limitations, we turn to self-supervised learning. Instead of predicting GO terms from domains, we aim to correlating them. Using a data-driven protocol, we learn the protein domain and GO terms co-occurrence relationships in the form of conditional probabilities p(GOj|domaini), i.e., the probability that a protein containing domaini performs the function GOj . At the core of our method is a simple yet effective neural network, what we call DomainGO prob. Fig. 2a shows the network architecture. This model maps the domains and GO terms into two separate embedding spaces and strives to establish correlation between them. Instead of directly regulating the dot product or similarity of the two embeddings, we apply Hadamard product, which is element-wise multiplication between two embedding vectors, i.e, the individual components of the dot product. This enables us to exploit higher-dimensional relationships between the two embeddings, in contrast to an overall aggregation obtained from dot product. We intentionally kept the network architecture shallow and simple, so that the functional information is contained within the actual embeddings and not in the auxiliary network layers facilitating the learning. While it was possible to design a more complex network architectures with deeper and wider layers, in those cases, the networks tended to perform the heavy lifting, making the embeddings less informative in the process. As a result, the embeddings of the domains become functionally informed, through optimizing the domain-function conditional joint probability prediction.
Fig. 2b explains how we developed the dataset to compute domain-GO associations with the network in Fig. 2a. We collected GO functional annotations of proteins from a database, including Swiss-Prot and extracted the protein domains using InterPro Scan. Based on the sets of obtained GO terms and domains, we compute the aforementioned conditional probability p(GOj|domaini) through a conventional counting-based approach.
Fig. 2c shows how the computed DomainGOProb is used for predicting function of a query protein.. During inference, from a query protein we identify domains using InterPro Scan and obtain the corresponding domain embeddings. Upon averaging the domain embeddings a functionally informed representation for the protein can be devised, and a supervised classifier can be applied to predict the functions of the protein. We name this overall protocol Domain-PFP.
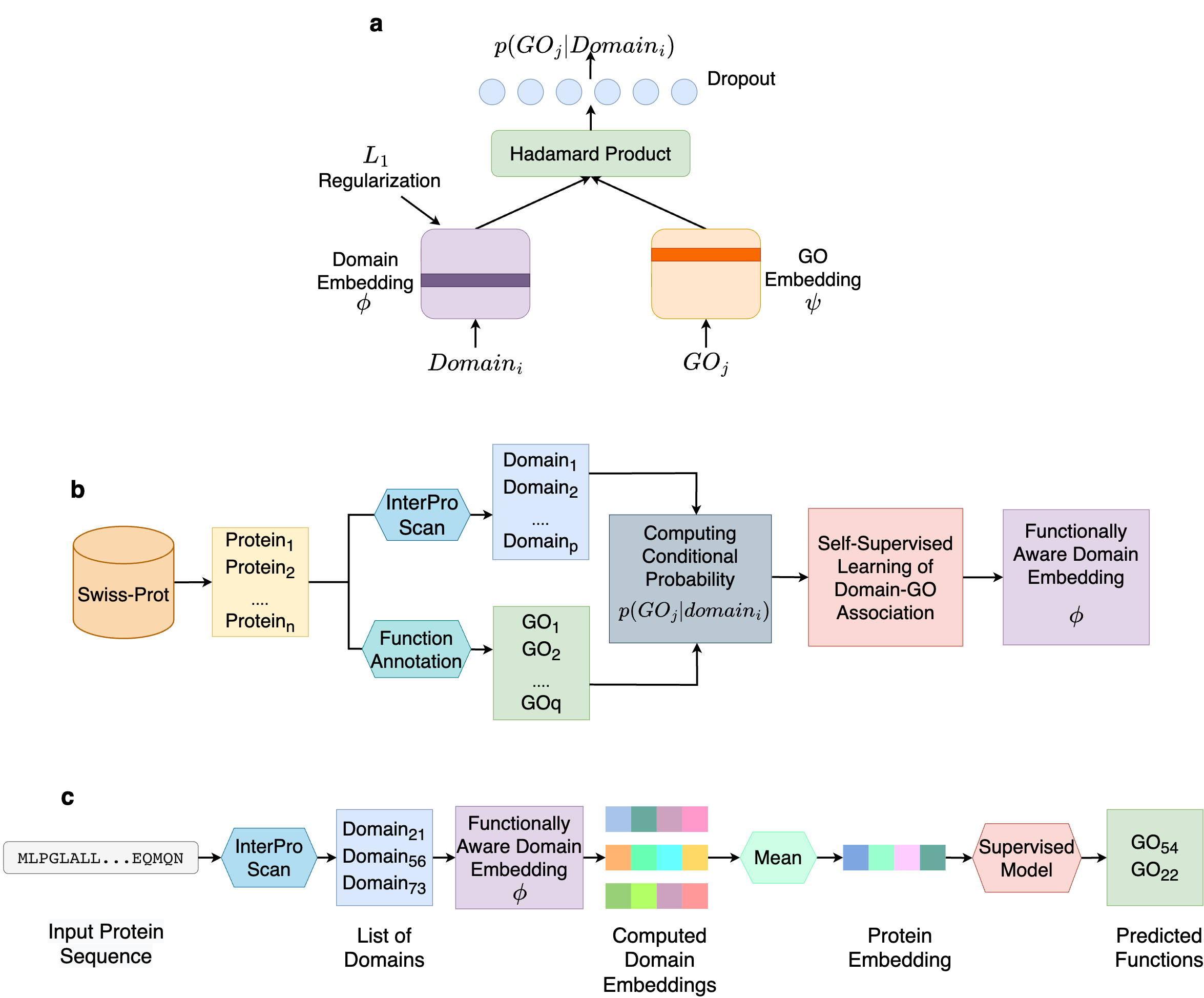
Figure 2. Overview of Domain-PFP. a, the network architecture used for self-supervised learning of domain embeddings. b, the overall pipeline of learning the functionally aware domain embeddings. c, the steps of computing the embeddings of a protein and inferring the functions.
Fig. 3 shows how protein embeddings in DomainPFP (Fig. 2c) correlates with functional similarity, measured with funSim Score. As shown, proteins at close proximity in the embedding space have a high funSim score greater than 0.4, which gradually falls as we consider proteins that are further apart. This score sharply falls when the embedding distance between proteins exceeds 5 unit.
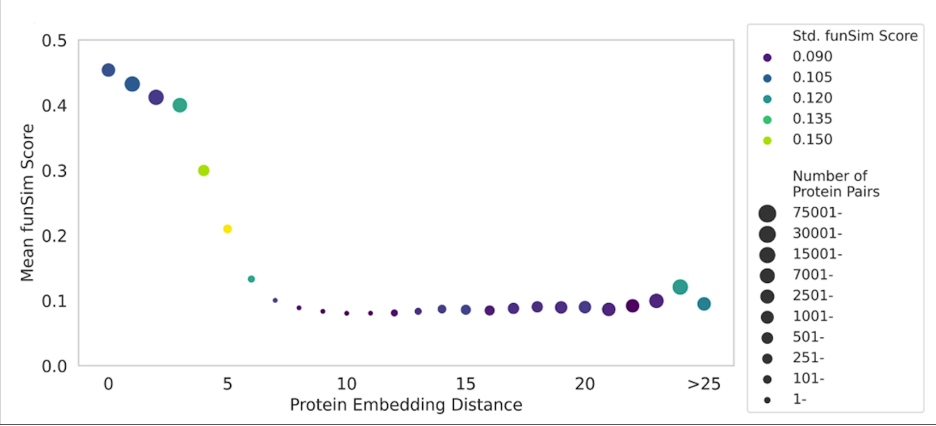
Figure 3. Correlation between the distance of protein embeddings and functional similarity.
In our work, we also showed that that our developed embedding contains much richer functional information than state-of-the-art protein language models, which are orders of magnitudes more complex and trained on massive volumes of sequences compared to our model. We have achieved up to 20% higher weighted F1-Score (a score for function prediction accuracy) for rare and specific GO terms through a simple linear classifier, which indicates that our representation is so much functionally informed that it is almost linearly separable.
Finally, we compared Domain-PFP (Fig. 2c) with state-of-the-art function prediction methods. These methods involve complex network architectures for instance, convolutional neural networks, graph neural networks, and transformers. We have observed that Domain-PFP, outperforms all other existing methods with similar level of provided information. Domain-PFP substantially improved by incorporating complementary data sources, such as BLAST and Protein-Protein Interaction (PPI). Moreover, our method demonstrated competence in CAFA3 evaluation along with structure-based function prediction datasets.
To contribute to protein function prediction research, we have released our source codes at https://github.com/kiharalab/Domain-PFP, and we have also developed an online platform for easy access at https://bit.ly/domain-pfp-colab. If you have any questions or potential ideas to further improve Domain-PFP, please contact Daisuke Kihara (dkihara@purdue.edu).
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in