DOPAL initiates αSynuclein-dependent impaired proteostasis and degeneration of neuronal projections in Parkinson’s disease
Published in Neuroscience

Aging is among the prominent pathological factors leading to Parkinson’s disease (PD), as it represents the greatest challenge for upholding efficient degradative pathways, thus altering neuronal protein quality control. At striatal terminals, the misfolding and aggregation of αSynuclein (αSyn) constitutes a driving factor in synaptic derangement.
Dopamine dyshomeostasis is known to contribute to nigrostriatal neuronal dysfunction by affecting several intersected pathways, which lead to a negative loop of mitochondrial and lysosomal dysfunction, and protein aggregation.
More recently, the unique interplay between the reactive dopamine catabolite 3,4-dihydroxyphenyacetaldehyde (DOPAL) and αSyn has gained attention within PD research community. The primary site of DOPAL burden is the pre-synaptic terminals, where its buildup is favored by a combination of defective dopamine storage in synaptic vesicles, increased MAO activity with aging and decreased DOPAL detoxification by ALDHs (Figure 1).
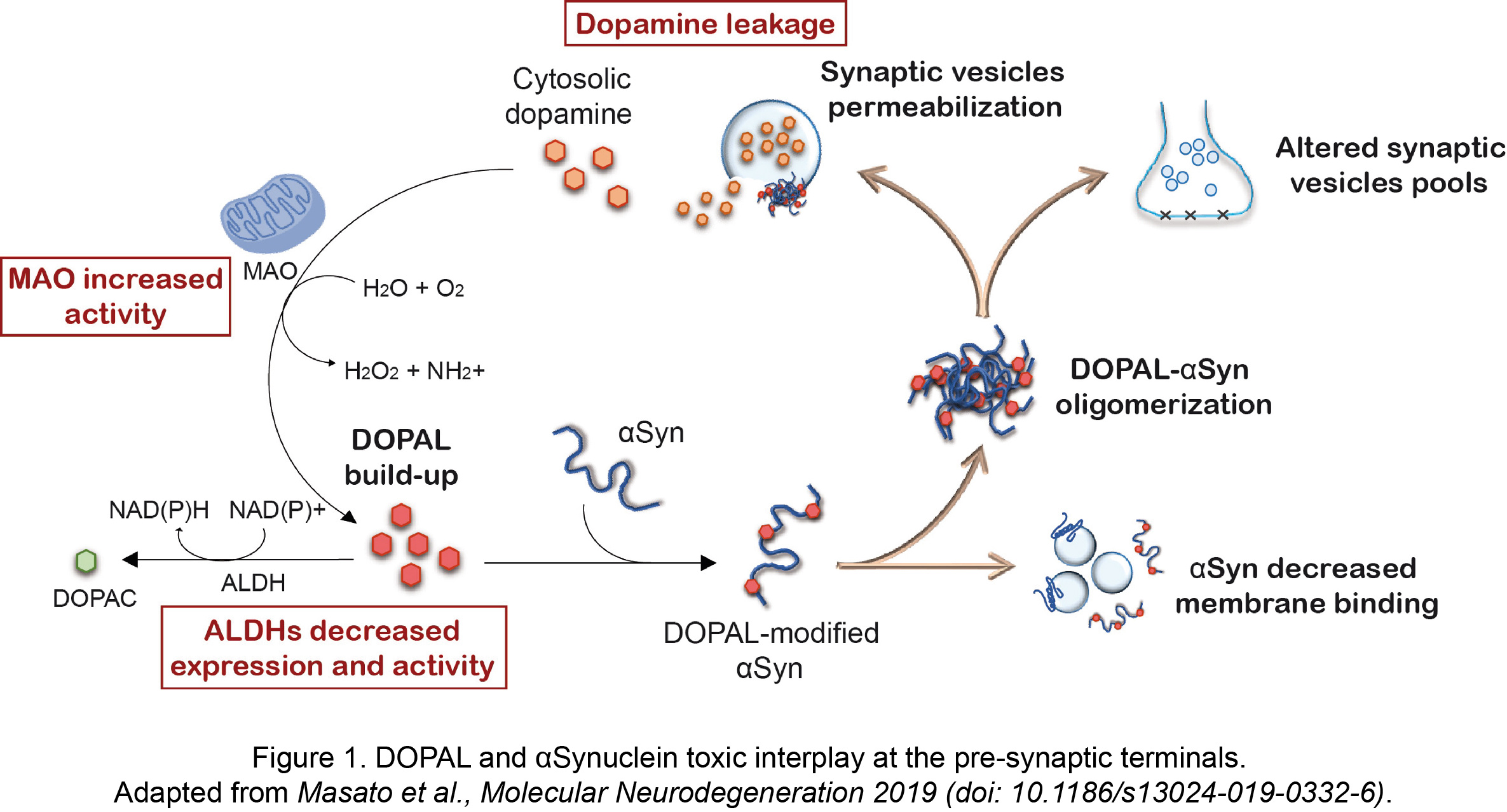
We previously demonstrated a functional consequence of DOPAL buildup at the pre-synaptic region, which induces a redistribution of synaptic vesicle pools in primary neuronal cultures (Plotegher et al., Scientific Reports 2017, doi: 10.1038/srep40699). This was linked to the generation of DOPAL-triggered annular-shaped αSyn off-pathway oligomers that were able to form pores on vesicles membrane (Figure 1). As in other studies, we showed that DOPAL covalently modifies lysines on αSyn sequence in a Schiff-base reaction between the primary amines and the aldehyde, with a higher and more specific reactivity than catecholamines.
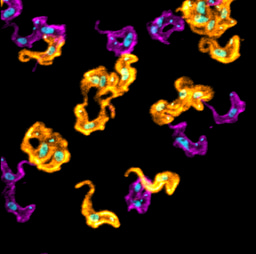
In our recent article published in npj Parkinson’s Disease, we untangled the impact of DOPAL buildup on neuronal homeostasis, in the light of DOPAL as trigger of αSyn-mediated neurotoxicity. By combining biochemical, cellular and imaging techniques, we demonstrated a DOPAL-induced αSyn accumulation among neuronal compartments and impaired αSyn clearance in DOPAL-treated primary neuronal cultures (Figure 2). Specifically, the correlated light and electron microscopy (CLEM) studies highlighted a DOPAL-stimulated αSyn accumulation along the endo-lysosomal pathway, with αSyn overload in lysosomal-like compartments, increased packing of intraluminal vesicles within multi-vesicular bodies and αSyn release via exosomes (Figure 2).
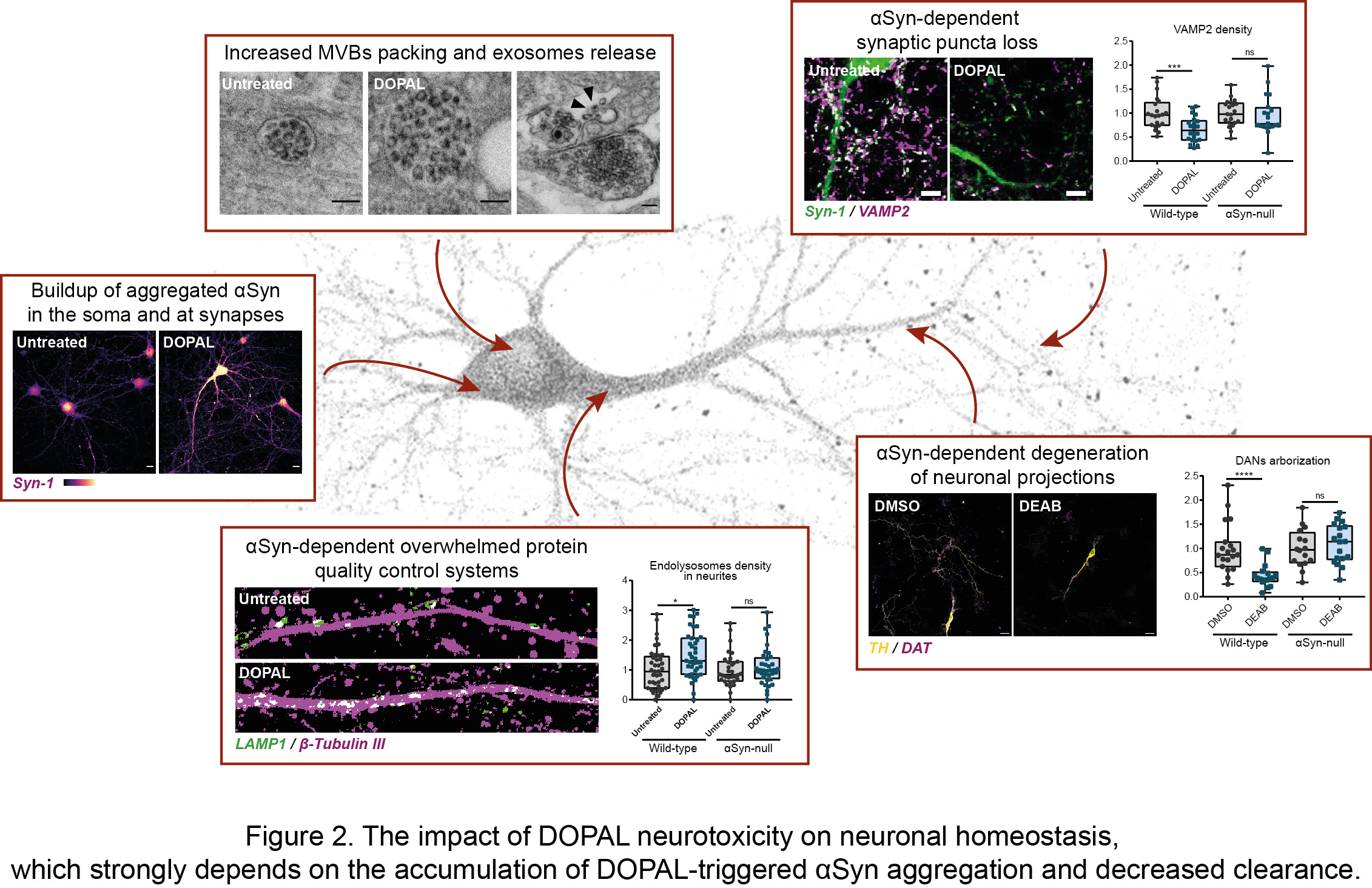
We discriminated the impact of DOPAL-triggered αSynuclein pathology on the soma and neurites, revealing more pronounced alterations of synaptic integrity, challenged protein quality control machineries in the neuronal projections and a reduced axonal arborization (Figure 2). Interestingly, the comparison of wild-type and αSyn-null neurons, we showed that DOPAL neurotoxicity strongly depends on its interaction with αSyn. Overall, our results can be coherently framed within the current view on PD etiopathogenesis of the dying-back hypothesis, for which we identified a plausible molecular mechanism.
Moreover, these observations were substantiated in in vivo models of defective DOPAL detoxification, namely the double knock-out mouse for both the cytosolic and mitochondrial ALDH enzymes (Aldh1a1-/-/Aldh2-/-) and zebrafish larvae exposed to the irreversible ALDH inhibitor and anti-abuse drug disulfiram (Figure 3A). Both animal models exhibited αSyn accumulation, dopaminergic neuron loss and impaired motor phenotype. In particular, the exposure to disulfiram resulted in a dose-dependent decreased swimming behavior in the zebrafish larvae, with a striking positive correlation with the loss of dopaminergic neurons in the diencephalon cluster, which corresponds to the nigrostriatal system in these animals (Figure 3B). Of note, the CRISPR-knockout of two Synuclein isoforms in zebrafish, sncga and sncb, revealed a rescue in the motor performance in the disulfiram-exposed sncga-KO zebrafish larvae. Hence, our data disclose a novel pathological interplay between αSyn and DOPAL as a key molecular mechanism of enhanced dopaminergic neuron vulnerability as an early events of PD.
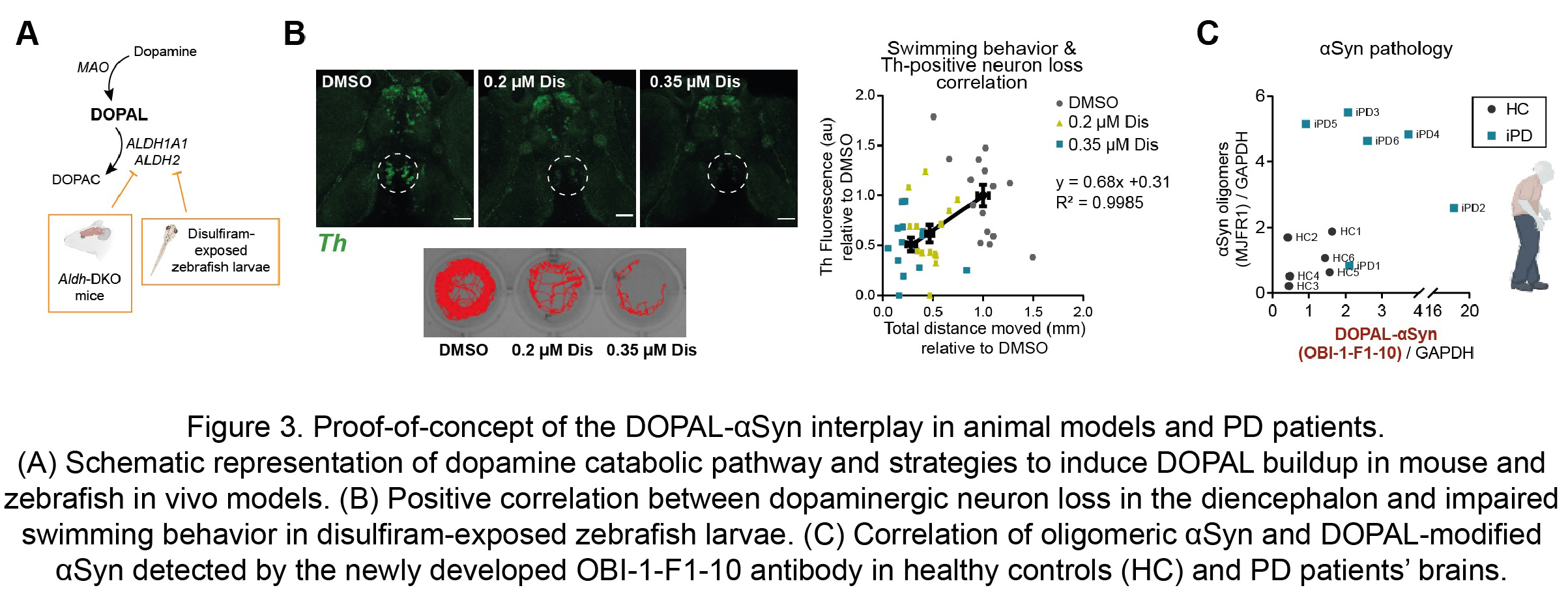
The mechanism proposed here acquires relevance if one considers its confinement within dopaminergic nigrostriatal neurons, at the early stages of PD pathology. For this reason, we put great effort in the development of a specific antibody against DOPAL-modified αSynuclein which allowed to confirm its presence of higher levels in human striatal tissues form idiopathic PD patients as compared to age-matched healthy controls (Figure 3C).
In our view, this work may provide a valuable step forward for the development of new biomarkers specifically aimed at revealing the dopaminergic imbalance and the generation and spreading of toxic DOPAL-αSynuclein species. By extension, this will contribute to the design of novel patient stratification strategies and, possibly, early therapeutic interventions that may act as disease-modifiers for PD.
Follow the Topic
-
npj Parkinson's Disease
This journal publishes original basic science, translational and clinical research related to Parkinson's disease, including anatomy, etiology, genetics, cellular and molecular physiology, neurophysiology, epidemiology and therapeutic development and treatments.

Your space to connect: The Psychedelics Hub
A new Communities’ space to connect, collaborate, and explore research on Psychotherapy, Clinical Psychology, and Neuroscience!
Continue reading announcementRelated Collections
With Collections, you can get published faster and increase your visibility.
The neuroimmune-axis and ageing in Parkinson’s Disease
Publishing Model: Open Access
Deadline: Jul 15, 2026
Cognition - preclinical models, and preclinical unmet need
Publishing Model: Open Access
Deadline: Jul 27, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in