Dos & don’ts for Capturing Multivalent Peptide Interactions in Real-time
Published in Protocols & Methods
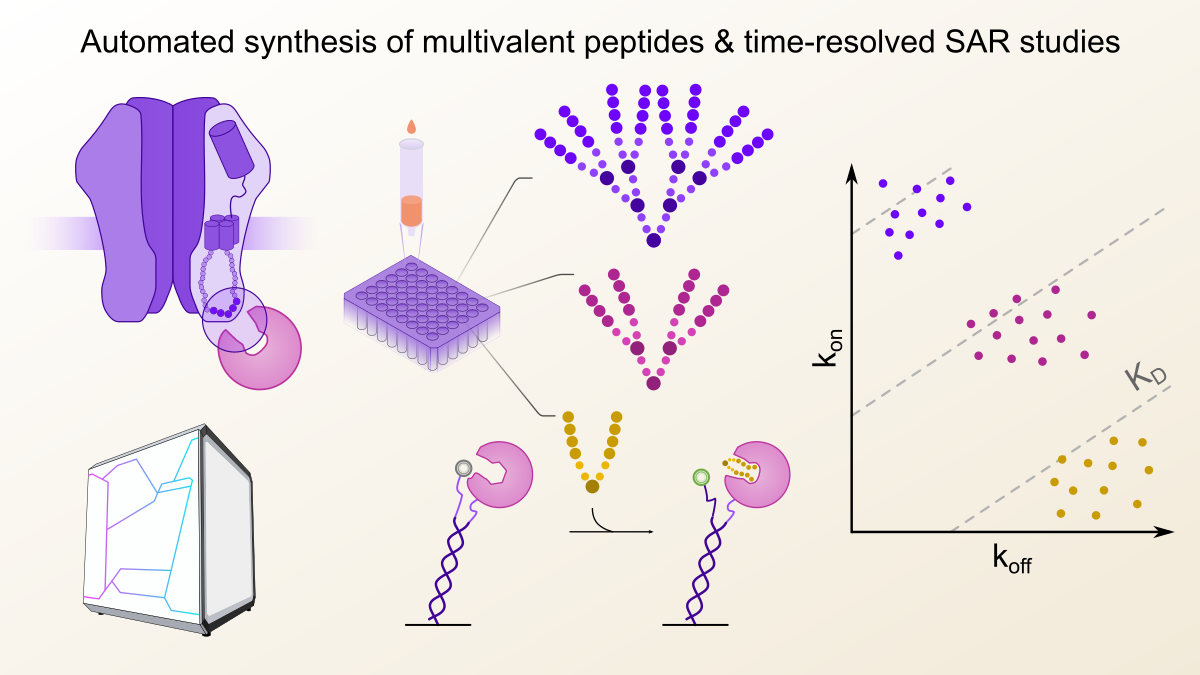

Interactions between different proteins in the human body, also referred to as protein-protein interactions (PPIs), are increasingly recognized as attractive drug targets and probing their function with specific inhibitors dramatically advanced our understanding about their contribution to physiology and pathology. Designing such inhibitors is not an easy task however, as firstly, it requires detailed biophysical knowledge about the PPI to be addressed, in order to construct binders or inhibitors with high specificity and binding strength, also referred to as binding affinity. At the start of this project, my goal was to construct multivalent peptides that would inhibit gephyrin, the master regulator of inhibitory postsynapses, expanding on earlier work from my labmates that demonstrated superior performance of dimeric binders – both in terms of inhibition potency but also specific targeting in the context of cells and even tissues 1.
This example and many others from the literature showed that multivalent binders had an especially notable binding affinity advantage due to a lower off-rate, or in other words, a prolonged half-life of the interaction, before the inhibitor dissociated from its target 2. I was surprised to find that in almost all cases the authors did not conduct a thorough and systematic screen of different valencies, linker types or complex multimeric architectures. Indeed, using conventional approaches it would take more than a single PhD worth of time to synthesize the necessary number of different compounds, let alone their precise thermodynamic and kinetic evaluation.
To this end, I was looking for methodologies that allowed an exhaustive screening, spanning over 100 different multivalent architectures. My hope was that such a large dataset may allow to draw general principles about the relationship between the multivalent peptide architecture and their binding affinity as well as kinetics. The readout should further enable to directly monitor peptide-protein binding in real time, and thus resolve the binding event (on-rate) and dissociation phase (off-rate) separately even when comparably small amounts of the peptide binders were available.
With these prerequisites in mind, I first explored Biolayer Interferometry (BLI) since it works without direct fluorescent labelling, which would ensure the functional integrity of the target protein and does generally not require a large amount of target protein for sensor functionalization. Indeed, in this case BLI consumed around 10-fold less target protein per affinity determination compared to the Isothermal Titration Calorimetry (ITC) measurements, which are often considered the gold standard of biophysical binding affinity determination. My first pilot experiments were quite encouraging, since the affinity of a series of dimeric peptides binding to gephyrin could be reliably determined, although with a non-ideal signal-to-noise ratio. However, as I attempted to measure the higher-architecture peptides, that contained a total of four or even eight binding epitopes, I noticed a strange behaviour during the dissociation phase. Specifically, the off-rate of the tetrameric and octameric series could not be resolved, regardless of architecture. After a long while of troubleshooting and testing a variety of modifications to the BLI binding assay, we concluded that we had encountered an inherent limitation of the BLI methodology, which had already been described in the literature for other immobilisation-based technologies such as surface plasmon resonance (SPR) 3. We hypothesized that due to the close proximity of the immobilized proteins on the sensor surface, the higher architecture peptides got irreversibly entangled, unable to dissociate again, which made an affinity determination impossible (Figure 1). For both SPR and BLI the textbook answer would be to reduce the immobilisation density which however in my case just let to loss of signal over noise. This of course was a bit of a disappointment, but once again underlined, that sometimes it is necessary to think outside the box to capture certain interactions. One thing was clear though: we were in need of a biophysical technique that would allow to immobilize the target protein at lower density, ideally in a defined distance and at the same time provide high signal-to noise ratios, regardless of ligand size.
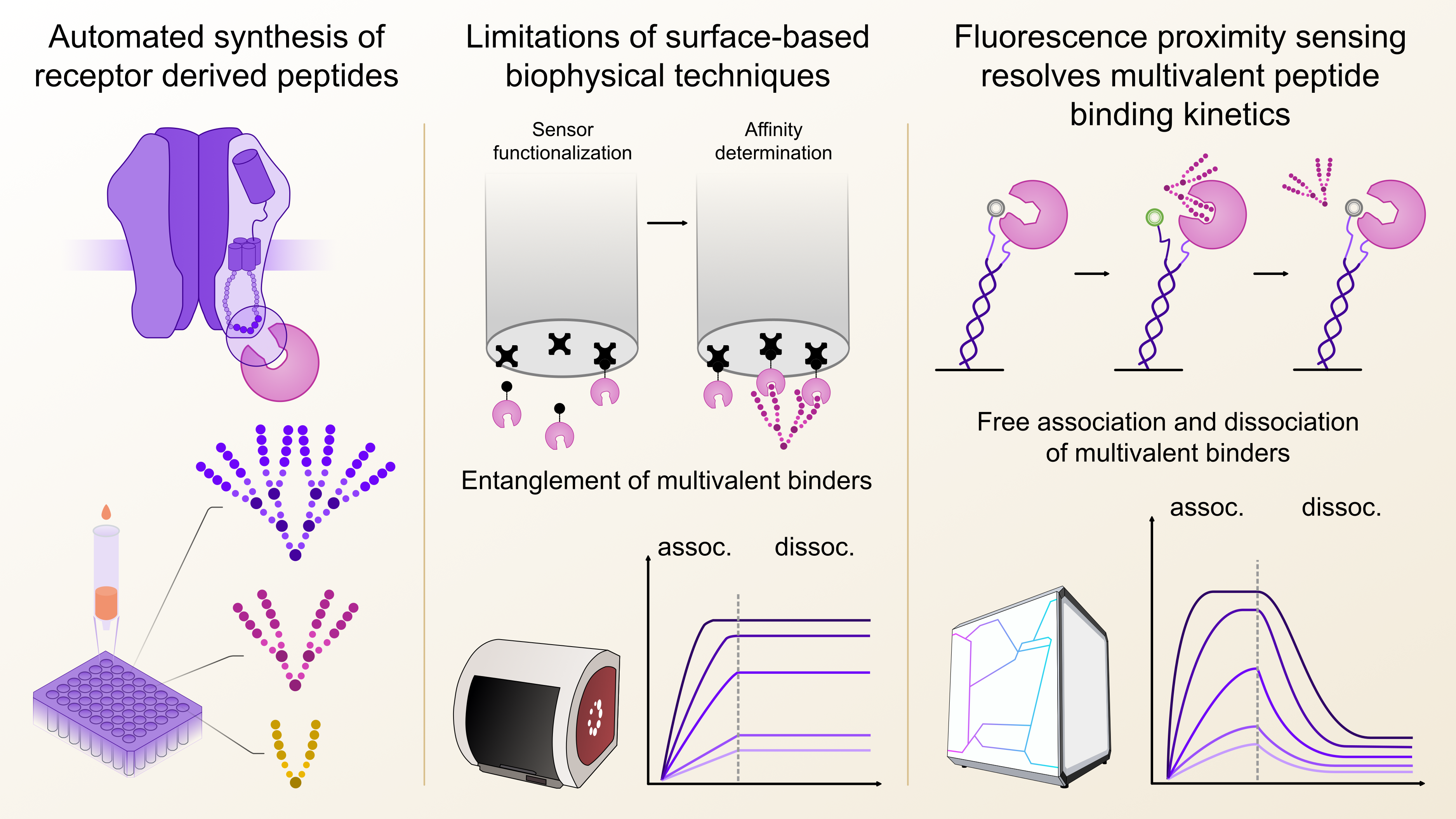
Figure 1: Fluorescence proximity sensing overcomes the limitations of surface-based kinetic biophysical techniques. Kinetic profiling of multivalent peptides from small scaled automated synthesis with biolayer interferometry was hindered by irreversible entanglement of the multivalent binders on the sensor surface. Especially the off-rate or dissociation (dissoc.) could not be determined. In fluorsecence proximity sensing, this limitation was overcome by immobilizing the target protein at a defined distance.
Luckily, around the same time, we got in contact with Dynamic Biosensors GmbH (Munich/DE), and their switchSENSE® technology that works by immobilizing target proteins at distances of around 30 nm using DNA strands, which prompted us to explore whether this approach could also prevent the complex re-binding effects we observed in BLI. In addition, since my readout would be based on the displacement of a fluorophore, I expected that the signal amplitude would not dependent on the protein loading density, unlike in the BLI measurements.
Alice Soldà, an expert in setting up the heliX® system, quickly demonstrated that it was possible to resolve on- and off-rates not only for dimeric but also tetrameric and even octameric peptides. After some more assay optimization, such as selecting the best possible fluorophore for detection and setting up control experiments to check the functional and structural integrity of the target protein, we went on to screen over 100 peptides, which due to the large degree of automated processes in the heliX® system, could be done essentially overnight.
Considering the size and complexity of the dataset obtained, I was curious what critical details were hidden within but difficult to recognise for the human perception alone. This is why I contacted Sebastian Spänig from the Dominik Heider lab, experts in the field of biomedical machine learning algorithms. Subjecting the obtained dataset to his custom algorithm revealed the most critical characteristics of the multivalent peptides that govern binding affinity. Most importantly, we found that introducing higher flexibility with polyethylene glycol linking units close to the binding epitopes resulted in higher binding affinities than introducing flexibility in the core of the multimer. To our surprise, the higher affinity of tetramers and octamers did not mainly arise from an increased half-life of the interaction, but rather from an increase in the on-rate, or in other terms, the speed at which the interaction was formed as the peptides were exposed to the target protein. Our hypothesis for this unexpected observation was that in our case, due to the dimeric nature of the target protein, any excess valency of the peptide binder would improve the formation of the complex due to optimal pre-orientation rather than stabilizing it.
The obtained datasets were orthogonally validated by Nanotemper GmbH (Munich, DE), which commercialize the Dianthus® system for in-solution affinity measurements that leverage the temperature related intensity change of fluorescent dyes. Here, we could validate the affinity of selected multimers that were in good alignment with our in-house ITC measurements. Finally, I tested selected candidate multimers and found that the best in vitro candidate also displays a high potency for neutralising the endogenous target protein ex vivo.
In summary, our work highlights limitations and advantages of specific biophysical technologies to evaluate multivalent binders. I am thankful for all the help and input and hope my efforts inspire others to systematically explore their own multivalent binders to provide novel chemical biology tools or explore new pharmaceutical strategies!
Find us on Twitter @SchulteClemens and @HMaricLab
1 Khayenko, V. et al. A Versatile Synthetic Affinity Probe Reveals Inhibitory Synapse Ultrastructure and Brain Connectivity. Angewandte Chemie International Edition, e202202078 (2022).
2 Wooldridge, L. et al. Tricks with tetramers: how to get the most from multimeric peptide–MHC. Immunology 126, 147-164 (2009).
3 Errington, W. J., Bruncsics, B. & Sarkar, C. A. Mechanisms of noncanonical binding dynamics in multivalent protein–protein interactions. Proceedings of the National Academy of Sciences 116, 25659-25667 (2019).
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in