Dual Gold Catalysis with Enzymes?
Published in Chemistry
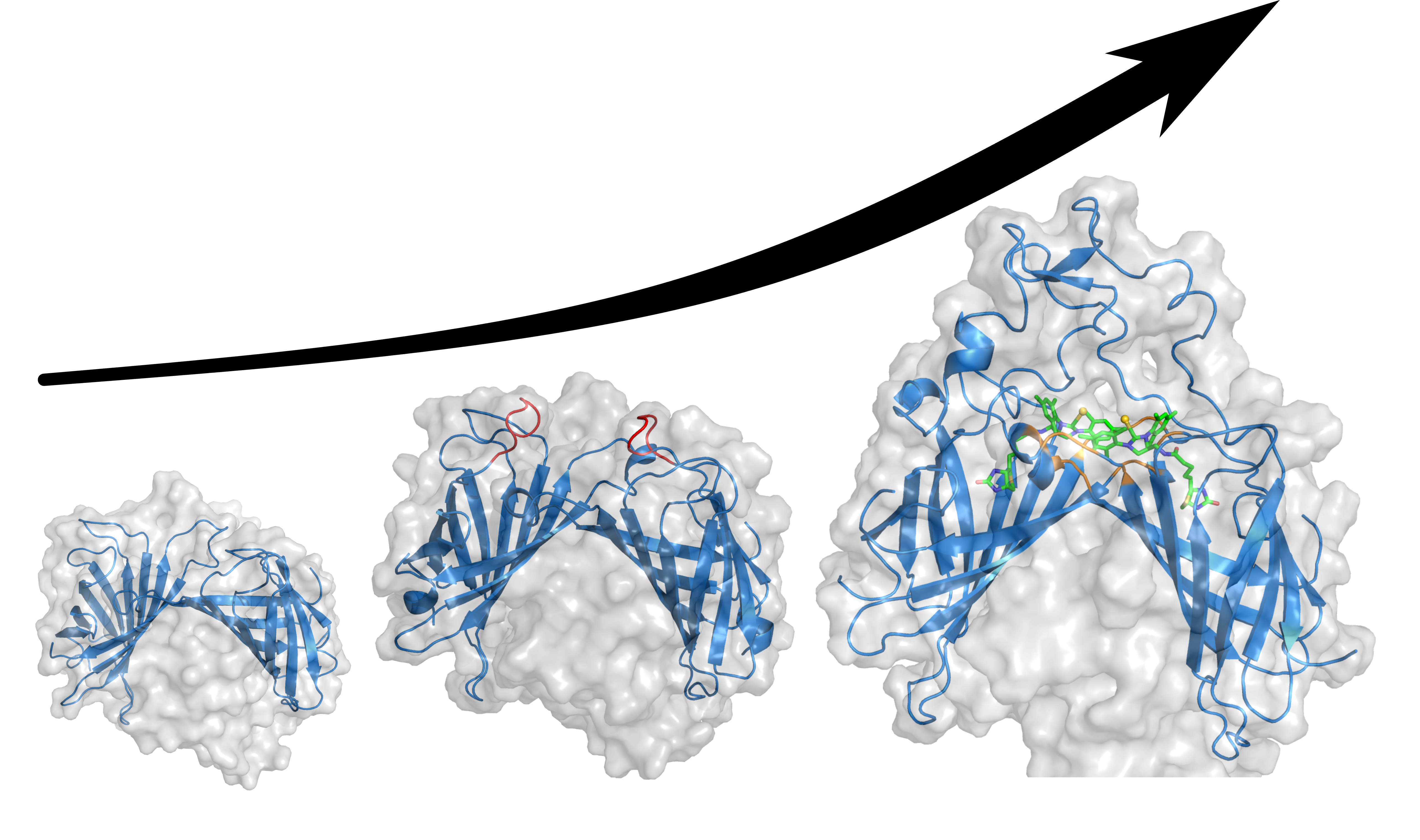

Enzymes are nature’s best chemists. They catalyze the conversion of substrates to a desired product, with exquisite selectivity and in a complex cellular environment. Artificial metalloenzymes (ArMs) offer an attractive means to capitalize on some of these properties and to apply them to organometallic catalysis. ArMs are assembled by localizing an artificial metallocofactor within a protein scaffold. Over the years, several proteins have proven particularly versatile for the assembly of ArMs. These include: carbonic anhydrase, hemoproteins, prolyl oligopeptidase, lactococcal multiresistance regulator, four helix-bundles, nitrobindin, human serum albumin, streptavidin (Sav), etc. Inspired by a visionary publication by Wilson and Whitesides,1 our group has relied mostly on Sav to assemble Fe-, Pd-, Ru-, Ir-, Rh-, and most recently Au-based ArMs. Perhaps one of the most attractive features of these ArMs compared to traditional organometallic catalysts is that their performance can be optimized by directed evolution of the protein scaffold.2 We like to think that this strategy enables chemists to “endow organometallic catalysts with a genetic memory”.
Over the past twenty years, we have accumulated a fridge full of purified Sav mutants (> 100).3 We commonly refer to this as the “fridge”. Whenever a new cofactor is designed or a promising reaction is scrutinized, we evaluate a selected set of these readily available mutants. This “quick-and-dirty” effort yields precious insight on the suitability of genetic engineering to improve ArMs’ performance.
In contrast to most previous studies that typically aim at improving enantioselectivity,2 we set out to improve regioselectivity.4 With this goal in mind, we selected the Au-catalyzed hydroamination of ethynylurea 1, Figure 1. Depending on the activation mode of the alkyne by the catalyst, either an indole 2 (anti-Markovnikov product) or a quinazolinone 3 (Markovnikov product) are formed preferentially. While the quinazolinone 3 results from a single gold activation of the alkyne, the indole 2 requires a dual gold activation, whereby one Au is π-bound to the alkyne and the second gold is σ-bound to the deprotonated terminal alkyne. We hypothesized that, thanks to the unique topology of the biotin-binding vestibule, we may be able to position two biotinylated Au-cofactors in a propitious position, thus favoring a dual σ,π-activation of the terminal alkyne to afford the indole 2.
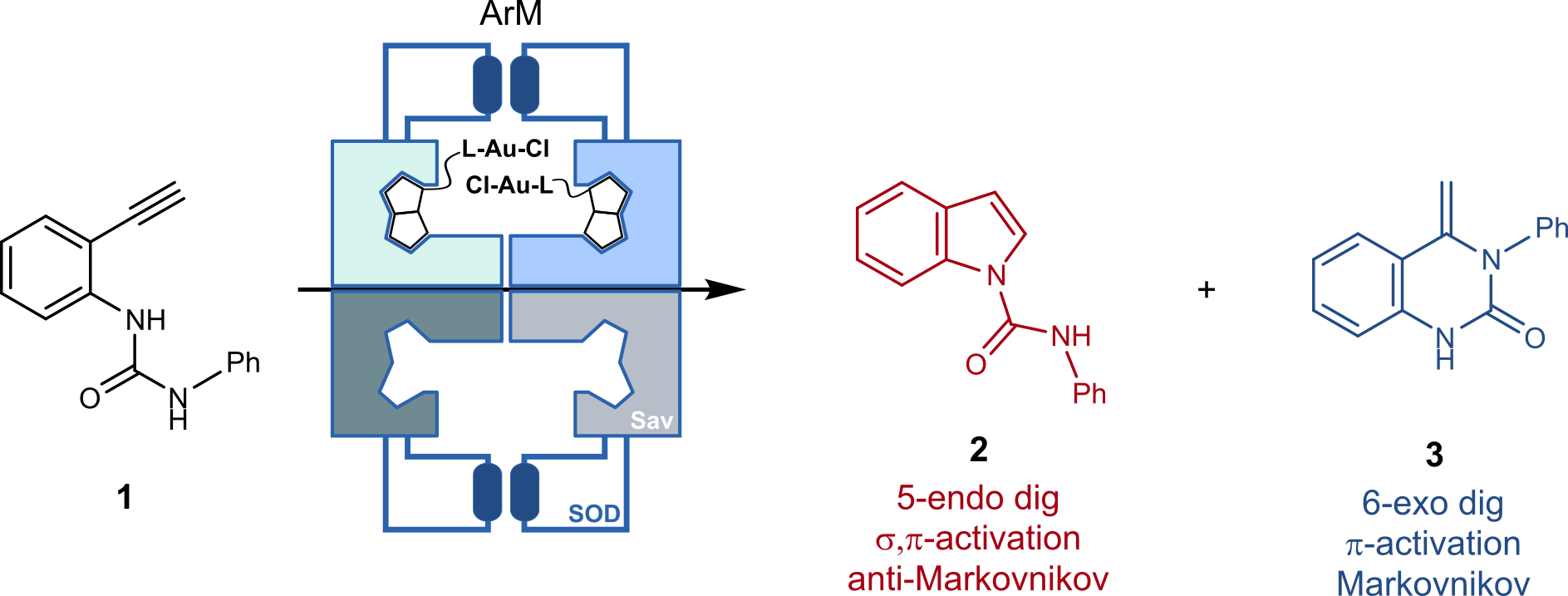
Figure 1 A biotinylated gold complex combined with a chimeric streptavidin (Sav) endowed with a hydrophobic lid borrowed from SuperOxide Dismutase (SOD) catalyzes the regioselective hydroamination of substrate 1 to afford either the indole 2 or the quinazolinone 3 depending on the activation mode.
Although point mutations indeed allowed to improve the turnover number, the ratio of products 2/3 remained by in large constant, favoring the quinazolinone 3. To shift the regioselectivity, we set out to engineer Sav to shield the biotin-binding vestibule from the bulk, thus forcing two adjacent biotinylated Au-cofactors to adopt a close-lying pose, Figure 2 a. This computationally-assisted feat was achieved by engineering a Sav chimera equipped with a 35 amino acid hydrophobic lid, borrowed from SuperOxide Dismutase C (Sav-SOD hereafter).5 The resulting chimera could be recombinantly expressed in E. coli in high yield and, the biotin-binding affinity was not significantly affected. Gratifyingly, the artificial hydroaminase based on this engineered Sav-SOD scaffold significantly affected the indole 2/quinazolinone 3 ratio. Next, we applied directed evolution to further improve the regioselectivity in favor of the indole product 2. Starting from Sav-SOD, three rounds of directed evolution covering up to six different positions led to highly selective mutants for either products, Figure 2 b.
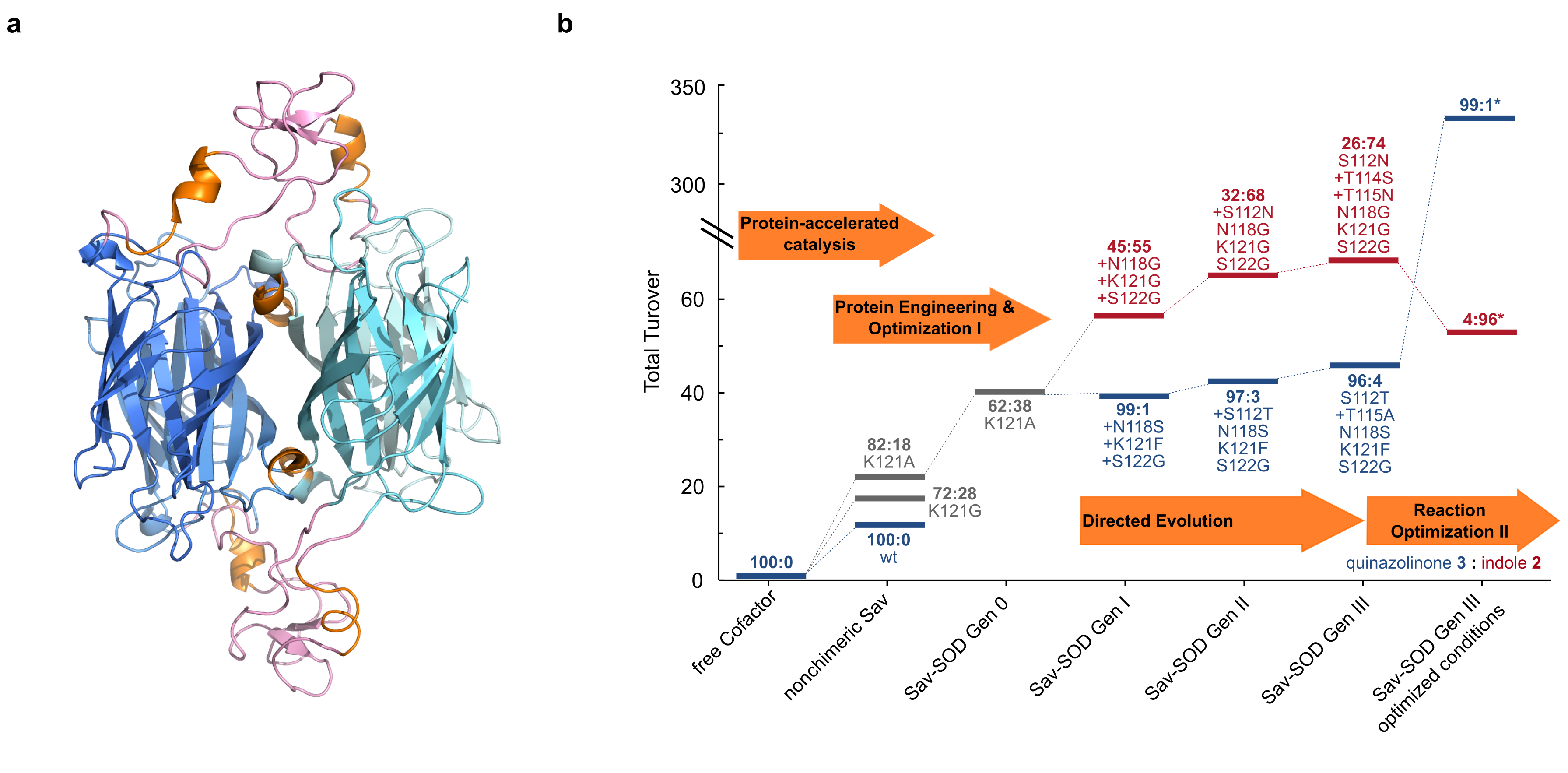
Figure 2 a Computed structure of the chimeric Sav-SOD resulting from a 200 ns MD simulation. b Evolutionary trajectories of Sav-SOD mutants for the hydroamination of substrate 1 to form either indole 2 or quinazolinone 3 (the product ratio 3:2 is displayed in bold).
Nature often relies on polynuclear metal cofactors to catalyze some of its most challenging reactions including: the oxygen evolving complex, nitrogenase, cytochrome c oxidase, soluble methane monooxygenase, etc. The realization of a dual-gold catalyzed hydroamination within an engineered Sav-SOD chimera highlights the versatility of ArMs to complement the enzyme’s repertoire with new-to-nature reactions.
For more information, please read our article in Nature Catalysis via https://www.nature.com/articles/s41929-021-00651-9
References:
- Wilson, M. E. & Whitesides, G. M. Conversion of a Protein to a Homogeneous Asymmetric Hydrogenation Catalyst by Site-Specific Modification with a Diphosphinerhodium(I) Moiety. J. Am. Chem. Soc. 100, 306–307 (1978).
- Schwizer, F. et al. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 118, 142–231 (2018).
- Vornholt, T. et al. Systematic engineering of artificial metalloenzymes for new-to-nature reactions. Sci. Adv. 7, 1–12 (2021).
- Gu, Y., Natoli, S. N., Liu, Z., Clark, D. S. & Hartwig, J. F. Site‐Selective Functionalization of (sp3) C−H Bonds Catalyzed by Artificial Metalloenzymes Containing an Iridium‐Porphyrin Cofactor. Angew. Chem. Int. Ed. 58, 13954–13960 (2019).
- Spagnolo, L. et al. Unique features of the sodC-encoded superoxide dismutase from Mycobacterium tuberculosis, a fully functional copper-containing enzyme lacking zinc in the active site. J. Biol. Chem. 279, 33447–33455 (2004).
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in