Efficient synthesis of multiple C–H bond functionalized unprotected alicyclic amines
Published in Chemistry
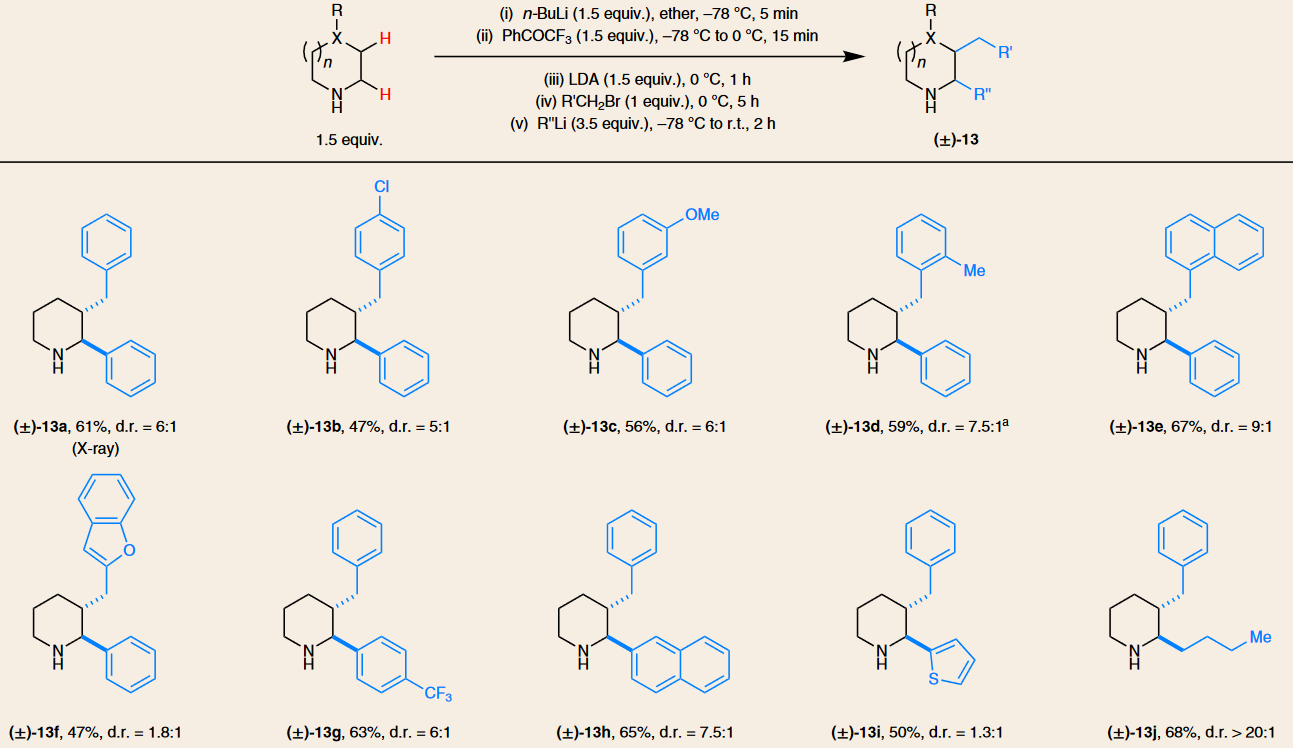

Inspired largely by the ubiquity of ring-functionalized cyclic amine structures in natural products and bioactive molecules, numerous methods have been developed to enable the selective C–H bond functionalization of cyclic amines. Previous studies largely focused on the most accessible α-C–H bonds. In contrast, functionalizations of more remote C–H bonds remain underdeveloped. In general, established methods to realize the goal of C–H bond functionalization at any ring position in cyclic amines are highly dependent on using structurally modified cyclic amines as starting materials, typically involving protection of the amine nitrogen atoms. These modifications of the starting materials inevitably add extra steps to the target transformation and can also limit the utility of the products for further processing. In addition, a large portion of reported methods in this area employ precious transition metals, which is something chemists often want to avoid.
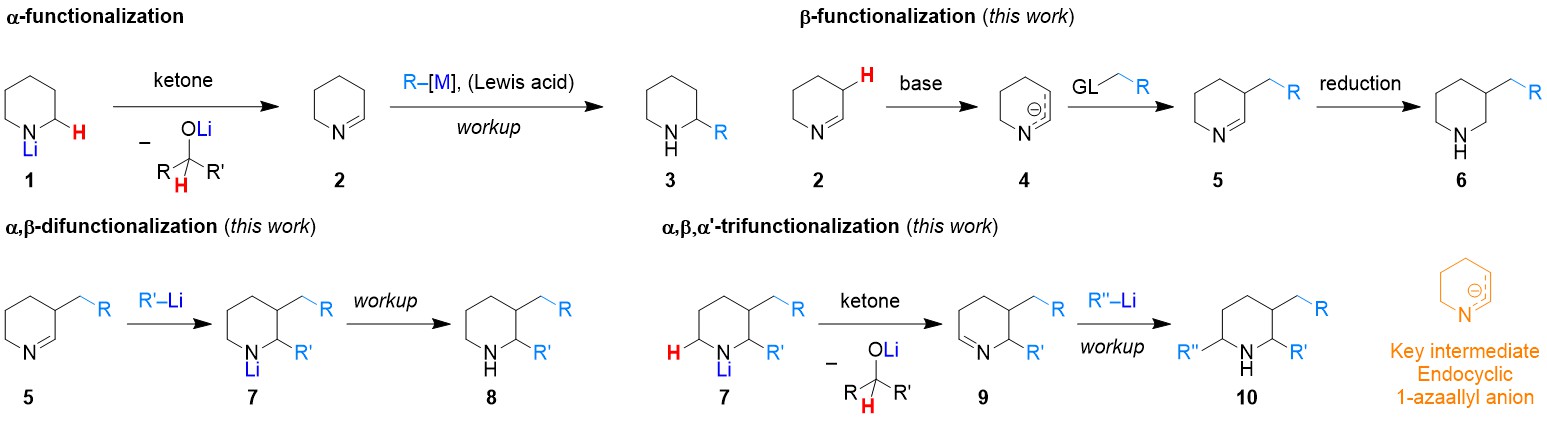
Our strategy for the β-C–H bond functionalization of cyclic amines utilizes unprotected alicyclic amines in the absence of transition metal catalysts. It is inspired by Wittig’s work on 3-alkylpyrrole formation from lithiated pyrrolidine and an imine, which was reported nearly forty years ago.[1] This precedent also provided part of the inspiration for our previous studies on the α-functionalization of unprotected alicyclic amines.[2] In the present work, deprotonated amines engage a sacrificial ketone oxidant to generate the corresponding cyclic imines, which are then deprotonated by LDA to form endocyclic 1-azaallyl anions as key intermediates. What we needed to accomplish is to identify proper conditions to enable the endocyclic 1-azaallyl anions (which are ambident nucleophiles) to selectively react with electrophiles at the C-terminus, followed by further processing of the resulting β-functionalized cyclic imines. The whole transformation consists of multiple stages that are conducted in one operation. Surprisingly, although their acyclic and exocyclic counterparts are well studied, endocyclic 1-azaallyl anions are exceptionally rare intermediates in chemical transformations. It was extremely gratifying to discover the optimal conditions under which unprotected alicyclic amines of various ring sizes readily undergo β-benzylation, allylation and alkylation. In the course of our study, we also found that endocyclic 1-azaally anions are mild nucleophiles in nature, since the reaction exhibits relatively broad functional group tolerance.
Extension of our strategy also enabled the sequential functionalization of up to three C–H bonds. The addition of three substituents at α-, β-, and α’-positions are orthogonal, regioselective and in most cases diastereoselective. To the best of our knowledge there is no other method that enables the rapid functionalization of multiple C–H bonds in unprotected alicyclic amines with only one isolation needed. We are hopeful that our method will become a useful tool for academic and industrial chemists.
If you want to learn more details about our method, please check our paper here: https://www.nature.com/article...
References:
[1] Wittig, G. & Hesse, A. Hydrid-übertragung von lithium-pyrrolidid auf azomethine. Liebigs Ann. Chem. 746, 174–184 (1971).
[2] Chen, W., Ma, L., Paul, A. & Seidel, D. Direct α-C–H bond functionalization of unprotected cyclic amines. Nat. Chem. 10, 165–169 (2018).
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in