Experiments inside a computer: polymer models to assess the performance of experimental methods for DNA spatial organization.
Published in Protocols & Methods

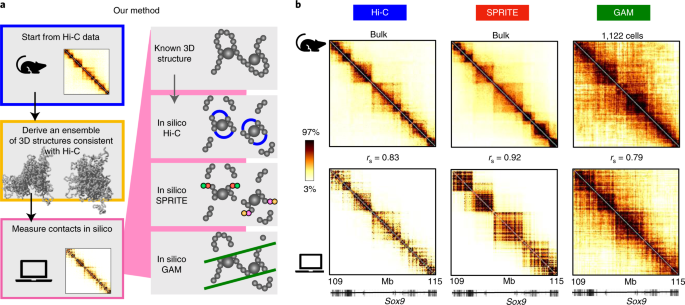
In mammal cells, DNA is far from randomly organized. The filaments of DNA in the nuclear volume orchestrate complex layers of loops, contacts and hierarchical architectures which are strictly connected to the functionality of the genome [1]. Therefore, the spatial organization of DNA is object of intense research. In the last decade, advanced experimental technologies have been invented to investigate the way DNA is shaped in cells, from super-resolution microscopy [2] to high throughput sequencing tools as Hi-C, GAM and SPRITE [3,4,5]. To make sense of the data collected by those powerful experiments, modelists interrogate polymer models which could unveil the molecular drivers of DNA architecture [6,7,8,9]. This is indeed the main research topic of our group.
As physicists interested in the three-dimensional (3D) organization of DNA in cells, we are used to testing polymer physics models over experimental data and then interrogating those models for novel insights. So, it was quite a challenge for us to tackle a project where that typical routine had to be reverted: using polymer models of DNA to benchmark experimental methods able to measure the 3D organization of the genome. To do so, we realized simulations of this kind of experiments on computers, with DNA represented by validated polymer models [7,10]. In this way, since all is known of polymer model structures, we could test the performance of various simulated experiments in many conditions, and in the controlled environment of a computer simulation. We thought of it as a virtual playground for experimental methods aimed at capturing the DNA organization in space.
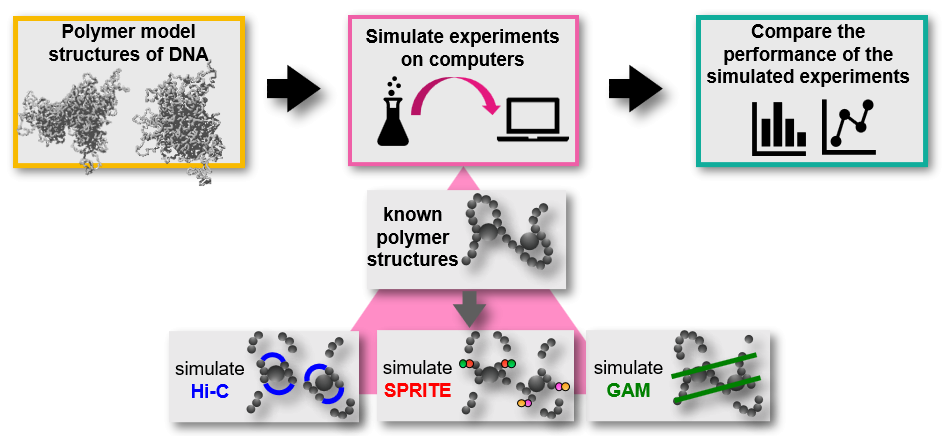
The first seeds of the project trace back to the contribution our group gave to the Ana Pombo’s lab in 2017 for the invention of the GAM technology [4], a method devised to investigate the spatial features of DNA. To do so, GAM recurs to a very different procedure than the one employed by Hi-C [3], the previously invented and much popular method to dissect DNA architecture genome-wide. So, a rigorous comparison between these two technologies was clearly in need, to understand their relative performances at given experimental conditions. The same necessity presented when also the SPRITE technology [5] was proposed in 2018, with features and protocol different from those of Hi-C and GAM. The questions we had in mind were several: which is the noise level affecting the output data of these technologies? How is their performance impacted by the experimental efficiency? At the same conditions, would these technologies return the same information on the spatial organization of DNA? And would this information faithfully capture the true spatial organization of the genome? Answering these questions experimentally is complex, as no clear benchmark can be used to assess the quality of the performance and, additionally, the amount of resources needed (money, biological samples, time etc.) would be significant. It was somewhere in 2018 that we thought we could try simulating a simplified version of Hi-C, GAM and SPRITE experiments on computers, where we could test them over known polymer structures in a controlled environment.
Our group had previously developed polymer models whose structures had proven to be a good proxy of real DNA configurations in cells [7,10], so we figured they could work nicely as benchmark for our virtual Hi-C, GAM and SPRITE methods. We devised algorithms that implement the main steps of the three technologies over polymer structures. For instance, one of the first steps of the Hi-C and SPRITE methods links together DNA sites in spatial proximity (the crosslinking of DNA), forming clusters of bonded DNA pieces; in our simulated version this is implemented by clusterizing polymer segments according to their spatial distances. As another example, in GAM a randomly oriented slice is extracted from the cellular nucleus by laser microdissection, so in the corresponding algorithm a random plane is produced in a spherical volume containing polymer structures.
With such algorithms up and running over our polymer models, we could think of the more disparate investigations. To mention few examples from our work, we studied how faithfully the outputs of Hi-C, GAM and SPRITE return the known spatial organization of the polymer structures, in different conditions. Also, we analyzed the level of noise affecting the outputs of Hi-C, GAM and SPRITE and, specifically, investigated its quantitative dependence on parameters of interest, as the experimental efficiency. From there, we elaborated a criterion to establish under which conditions the outputs of Hi-C, GAM and SPRITE are statistically robust. Interestingly, we found that the noise levels can be very different across Hi-C, GAM and SPRITE, due to the intrinsic differences in their protocols.
We believe model-based approaches as the one we present in our work can be useful for the design of novel experiments, orienting the choice of the most suited technology for a given task. We like to think of a future where more and more sophisticated computational experiments are enrolled on reconnaissance missions to guide the experimental efforts for the dissection of the spatial organization of DNA.
REFERENCES
- Pombo, A. & Dillon, N. Three-dimensional genome architecture: Players and mechanisms. Nat. Rev. Mol. Cell Biol. 16, 245–257 (2015).
- Bintu, B. et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science (80-. ). 362, eaau1783 (2018).
- Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (80-. ). 326, 289–293 (2009).
- Beagrie, R. A. et al. Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524 (2017).
- Quinodoz, S. A. et al. Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 174, 744-757.e24 (2018).
- Fudenberg, G. et al. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 15, 2038–2049 (2016).
- Conte, M. et al. Polymer physics indicates chromatin folding variability across single-cells results from state degeneracy in phase separation. Nat. Commun. 11, 3289 (2020).
- Jost, D., Carrivain, P., Cavalli, G. & Vaillant, C. Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res. 42, 9553–61 (2014).
- Brackley, C. A., Taylor, S., Papantonis, A., Cook, P. R. & Marenduzzo, D. Nonspecific bridging-induced attraction drives clustering of DNA-binding proteins and genome organization. Proc. Natl. Acad. Sci. U. S. A. 110, E3605–E3611 (2013).
- Chiariello, A. M., Annunziatella, C., Bianco, S., Esposito, A. & Nicodemi, M. Polymer physics of chromosome large-scale 3D organisation. Sci. Rep. 6, 29775 (2016).
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Oct 30, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in