Extending the small-molecule similarity principle to all levels of biology
Published in Bioengineering & Biotechnology

The chemical space is huge, with hundreds of billions of molecules synthetically accessible and a catalogue of commercial compounds that exceeds the hundred million, a few million of which are bound to be bioactive (i.e. able to interfere and modulate biological functions). Querying compounds in these databases differs greatly from querying proteins or genes, since small molecules are scarcely annotated, spread throughout various resources and cannot be split into functional groups. Often, the only way to approach the characterisation of a compound is to assume it will have the same activity as compounds with similar chemical structures. The so-called ‘similarity principle’ has been the driving force of drug discovery and, in one flavour or another, the calculation of compound similarities lays behind most of the methods used to chart and exploit the chemical space.
The ‘omics revolution’, and the broad release of small molecule bioactivity data, has led to the realisation that the similarity principle applies beyond chemical properties. For instance, we know that molecules with similar cell-sensitivity profiles or eliciting similar side-effects often have the same mechanism of action, even when their chemical structures appear to be unrelated. This suggests that ‘biological’ similarities offer an alternative means to navigate the chemical space, possibly unveiling non-obvious, clinically-relevant similarities between compounds. Unfortunately, though, there is no standard methodology to compare biological profiles as, for instance, 2D fingerprints or structural keys are used to compare the chemical structures of compounds.
We now present the Chemical Checker (CC), a resource that provides processed, harmonised and ready-to-use bioactivity signatures of small molecules. The CC divides data into five levels of increasing complexity, following the way we think of drug activity: a drug is often an organic molecule (Chemistry) that interacts with one or several protein receptors (Targets), triggering perturbations of biological pathways (Networks) and eliciting phenotypic outcomes that can be measured in cell-based assays (Cells) before delivery to patients (Clinics). Our resource contains data on the effects exerted by about 1M compounds in a wide range of biological settings, offering a rich portrait of the small molecule data available in the public domain, and opening an opportunity for making queries that would be otherwise impossible using chemical information alone. For instance, we used approved drugs and experimental compounds to revert Alzheimer’s disease transcriptional signatures in mutated SH-SY5Y cells, bringing key genes back to their physiological levels. We also looked for small molecules that could mimic the expression signatures resulting from the genetic inactivation of several cytokine receptors (e.g. IL2R, IL12R and EGFR), finding several compounds that could substitute antibody therapies against them. Besides, we envisage many other applications for the CC, such as the formulation of logical queries to prioritise drug repositioning and combination opportunities, based on desired bioactivity traits, or the use of bioactivity signatures to constrain generative models in de novo molecule design. Overall, the signature-based representation of compound bioactivities pushes the similarity principle beyond chemical properties, reaching various ambits of biology and enabling the right level of experimental detail at each step of the drug discovery pipeline.
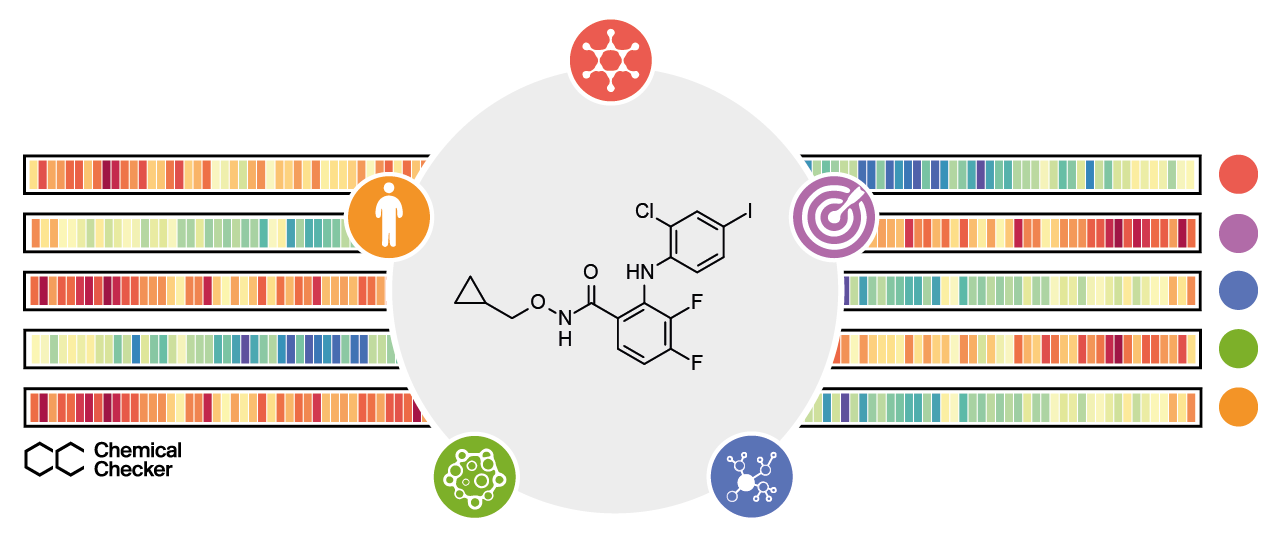
The CC signatures encapsulate small molecule bioactivities throughout five levels of increasing complexity: the chemical properties of compounds, targets and off-targets, perturbed biological networks, cell-based assays and, finally, clinical outcomes.
To facilitate access to our data, we built a web-based resource (CCweb; https://chemicalchecker.org) which, given a compound, allows to run similarity searches across the 25 available CC bioactivity spaces. The full code of the resource and CC signatures can be downloaded from the CCweb, or simply accessed via a programming interface.
You can read the full article here: https://www.nature.com/articles/s41587-020-0502-7
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in