Flu Junk
Published in Microbiology

Influenza viruses consist of little more than protein, lipid and eight strands of single-stranded RNA. However, in spite of their size, flu infections can result in a substantial disruption of our daily activities, severe pneumonia, or even death.
Milder forms of flu are common in infections with seasonal influenza strains, which are viruses that have adapted to humans and replicate well in human cells. Severe infections on the other hand are typically caused by highly pathogenic bird flu strains, such as H5N1 and H7N9. These strains occasionally “jump” over to humans, but do not spread well among people because they are poorly adapted to us.
Various experiments and evolutionary analyses have shown that avian influenza viruses can mutate or reassort with seasonal strains and acquire the ability to spread more efficiently. The most notorious example is the 1918 H1N1 pandemic virus, which causes an accumulation of fluids and white blood cells in our lungs, a shutdown of essential organs, blackening of toes and fingers, and eventually death by suffocation. In 1918, the virus killed about a 100 million people and there is substantial concern that if a similar virus emerged now, up to 6 times as many people could die.

Why avian viruses and the 1918 pandemic virus cause more severe disease than seasonal influenza strains is only partially understood. A critical factor appears to be the innate immune response. In infections with human adapted viruses this response tends to match the danger, clearing the virus without harming the host. However, in infections with avian influenza strains, the immune response can be disproportionate and hallmarked by high cytokine levels and extensive cell death.
The innate immune response detects replicating RNA viruses through cellular pathogen receptors. RIG-I is such a sensor and it can bind influenza virus RNA in vitro. However, what happens at a molecular level during infection is not so clear-cut, because the viral genome is normally shielded by viral proteins, such as the viral polymerase and nucleoprotein.
We became interested in what RIG-I “sees” during an avian influenza virus infection when we observed that truncated mini versions of the influenza virus genome triggered more cytokine expression than intermediate versions (also called defective interfering RNAs) or full-length flu RNA segments. Intrigued, we next found that mini flu RNAs were bound more effectively by RIG-I than longer RNAs in vivo. This is also seemed to fit the observation that mini flu RNAs were copied by the viral polymerase in the absence of other viral proteins, while longer RNAs were not. Potentially, mini flu RNAs were so small that only the polymerase could bind them, but other flu proteins could not, leaving mini flu RNAs more exposed to RIG-I, unlike the much longer flu genome.
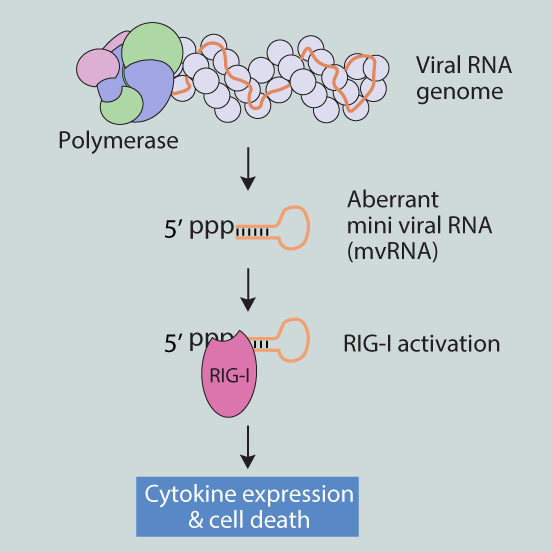
These observations raised a very interesting question. Could mini flu RNAs be generated during infection, for instance when the flu genome was copied incorrectly? To investigate this, we first took the polymerase of a virus that was well-adapted to human cells and tried to frustrate it. Immediately, mini flu RNAs appeared. Next, we did the same study with polymerases of the 1918 pandemic virus and a highly pathogenic H5N1 strain to see if this was a common feature. However, to our surprise, the enzymes of these two viruses made mini flu RNAs in human cells even when we did not try to frustrate them.
To verify whether the 1918 pandemic virus and avian influenza viruses also generated mini flu RNAs in vivo, we collaborated with colleagues at Rocky Mountains Laboratories, Erasmus Medical Centre, and University of Hong Kong. Analysis of the various samples by deep sequencing showed that there was a strong correlation between the presence of mini flu RNAs and the induction of innate immune signaling in lung tissue.
So, why do avian and pandemic viruses make these mini RNAs? Polymerases of avian influenza viruses differ from human-adapted polymerases at various amino acids. These may help the enzyme interact with key host factors or deal with the difference in body temperature between birds and mammals. We focused on a cluster of amino acids that could potentially help coordinate how RNA leaves the polymerase and becomes packaged by other the viral nucleoprotein. To test if mutations in this cluster were involved in mini flu RNA generation, we took the polymerase of the adapted virus again and changed the amino acids in the cluster to those of the 1918 influenza virus. Again, mini viral RNAs appeared, even under conditions where the original virus never made them and did not trigger any cytokine expression.
All our observations suggest that avian and pandemic viruses don’t copy themselves properly when they get inside human cells and that the resulting “junk” can be detected by our immune system. We think this is an exciting idea and an important step towards understanding what makes pandemic and avian influenza viruses so dangerous.

Our findings also raise new questions. Are mini flu RNAs beneficial to avian and pandemic viruses or are they just an unfortunate byproduct of a virus that is struggling to survive in a new host? How quickly do mini flu RNAs appear in an infection and can we negate their effects with drugs? Do more virulent seasonal flu strains, such as 2017’s H3N2 strain, make them too?
We are working hard on trying to answer these questions and hope that our observations also inspire other scientists. Together, we may be able to develop this interesting discovery into a method to identify dangerous viruses and combat them in the future.
Follow the Topic
-
Nature Microbiology
An online-only monthly journal interested in all aspects of microorganisms, be it their evolution, physiology and cell biology; their interactions with each other, with a host or with an environment; or their societal significance.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in