Generative AI and GPU-accelerated computing accelerate the discovery of future energy materials
Published in Materials, Computational Sciences, and Statistics

This blog presents a behind the scenes narrative on the work that we reported in our article A generative artificial intelligence framework based on a molecular diffusion model for the design of metal-organic frameworks for carbon capture, published in Communications Chemistry.
Why? The discovery of energy materials is of paramount importance to achieve time-critical clean energy goals. While I am a theoretical physicist and mathematician by training, and an avid AI practitioner, I understand the urgent need to develop novel AI approaches to tackle contemporary grand challenges in other domains, including materials science. Furthermore, I have met world class materials science researchers at Argonne National Laboratory, and their work has inspired me to learn more. To kickstart my training on AI for materials science discovery, I co-organized a Materials Science hackathon in 2022 with colleagues at the University of Chicago. This event attracted students from the University of Illinois, Northwestern University and the University of Chicago. A few of these students expressed a real interest in further developing their skills at the interface of AI, supercomputing, and materials science.
How? A few months after the hackathon, I had established a nice collaboration with Hyun Park and Ruijie Zhu, graduate students at the University of Illinois and Northwestern, respectively. In an attempt to translate lessons learned in my work on AI for physics, I worked with Hyun and Ruijie on the development of a minimal AI toolkit for materials science applications that would seamlessly work in modern computing environments. Thus, we assembled a suite of tools for hyperparameter optimization, distributed training, distributed and interpretable AI inference to provide researchers with a computational framework to model small molecules, inorganic crystals, and metal organic frameworks (MOFs) with state-of-the-art AI modes. The bonus of this work is that all the software needed to use these AI tools was designed to seamlessly work in leadership class supercomputers at the Argonne Leadership Computing Facility and the National Center for Supercomputing Applications. Though this last sentence may sound as an afterthought, deploying state-of-the-art AI models in supercomputers and then optimizing them to run at scale is very challenging. This work was published in End-to-end AI framework for interpretable prediction of molecular and crystal properties. By the end of this project I had developed a solid collaboration with Santanu Chaudhuri, and he encouraged his graduate student, Xiaoli Yan, to get involved in this work.
The breakthrough. I talked with my mentor at Argonne, Ian Foster, about the new direction of research I was exploring on generative AI applications for materials science, and he keenly supported this endeavor. His continued support and feedback changed the course of this project. I also believe that the recipe for success in this project is that the students involved had pretty diverse and complementary backgrounds. Hyun is a brilliant AI practitioner, Ruijie is an amazing scientific software developer, and Xiaoli has deep expertise in computational chemistry, with a very strong skill set in high performance computing. Through this project he has also developed a strong skill set in AI. With this deep pool of expertise, we posed the question: is it possible to design a robust generative AI method to assemble MOFs from the ground up? And if so, can we develop an open source, stand alone computational framework to screen and validate AI-generated MOFs for uniqueness, synthesizability, structural validity, stability and chemical consistency, and then quantify their carbon dioxide adsorption capacities? Over a period of six months, Hyun, Ruijie, Xiaoli and I developed, harnessed and adapted generative AI models and scientific software to painstakingly address these questions. With expert guidance from Ian Foster, Santanu Chaudhuri and Emad Tajkhorshid, we carefully assembled the pieces of this generative AI approach to accelerate the discovery of MOFs. Once we produced the key results of this work, we completed the manuscript in a couple of weeks. Refining the presentation and description of our methods and results took another couple of weeks. Precision matters a lot!
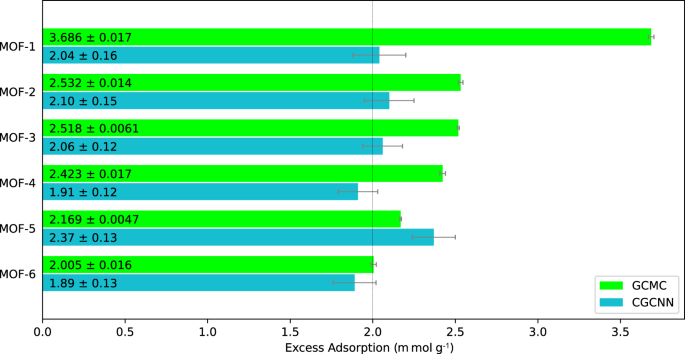
The reviews. The reviewers provided constructive and thoughtful comments, and as a result we added two important pieces to our computational framework, namely, a computational method to validate the geometry of our AI-predicted MOFs, and we also incorporated Grand Canonical Monte Carlo simulations to quantify the carbon dioxide adsorption capacities of our AI-generated MOFs.
Final thoughts. A vibrant, worldwide community of researchers is deeply involved in the development of disruptive AI methods for materials science. We can only imagine the future of this scientific revolution once AI is boosted with exascale computing. As it happened in physics several years ago, it is clear that AI for materials science is past the tipping point, a revolution is in the making. Thus the discovery of novel materials to achieve net zero, and other clean energy goals, is certainly feasible. As this AI program continues to elevate human knowledge, and transforms the practice of science, it will be important to remember that in any scientific endeavor the key challenges to overcome will not be computational, theoretical or experimental, but human.
I am Lead for Translational AI at Argonne National Laboratory, and CASE Senior Scientist at the Department of Computer Science at The University of Chicago. I have broad research interests at the interface of artificial intelligence, theoretical astrophysics, mathematics, extreme scale computing, and scientific visualization. I enjoy assembling and working with interdisciplinary teams driven by ideas that disrupt the state-of-the-art to enable sustained innovation.
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in