High-quality alligator gar chromosome-level genome aids in exploring its internal invasion mechanism
Published in Earth & Environment, Ecology & Evolution, and Zoology & Veterinary Science
The alligator gar invasion poses a serious ecological threat
The alligator gar is one of seven extant species of the ancient Lepisosteidae family. The slowly evolving genome of the gar fish has garnered increasing attention from scientific researchers in recent years. The draft genome of the alligator gar was used in a study examining the terrestrial transition of vertebrates from aquatic environments. It is noteworthy that recent reports highlighting the invasion of the alligator gar underscore the urgency of prioritizing its management efforts. Alligator gar is native to northern and central parts of the United States and Mexico. It has been distributed to numerous countries globally through the aquarium industry. Considered invasive in China, Singapore, Indonesia, Turkmenistan, and several other nations, it has been documented invading 47,287 locations across the planet. Initially detected in Baiyun Lake, Guangzhou, Guangdong Province, in February 2019, it subsequently expanded its range to various provinces including Hunan, Guangxi, Shandong, Sichuan, Qinghai, Jiangsu, and Yunnan. Despite the availability of a draft second-generation genome of the alligator gar, it largely limits the study of speciation and chromosome evolution.
Genome assembly at chromosome level
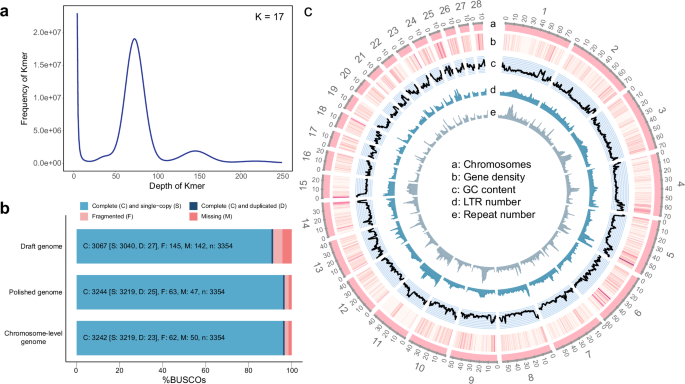
In this study, we generated a high contiguity, completeness, and accuracy genome assembly of alligator gar at chromosome level using Oxford Nanopore Technology and Hi-C sequencing techniques. The assembled genome is 1.05 Gb, with a contig N50 of 15.7 Mb, scaffold N50 of 56.8 Mb. The Hi-C sequences were further clustered and ordered into 28 pseudochromosomes(2n = 56, length from 10.2 Mb to 76.3 Mb). A sequence of ~323 Mb was annotated as a repeat element, constituting 30.91% of the genome. We predicted 19,103 protein-coding genes, of which 99.83% were functionally annotated.
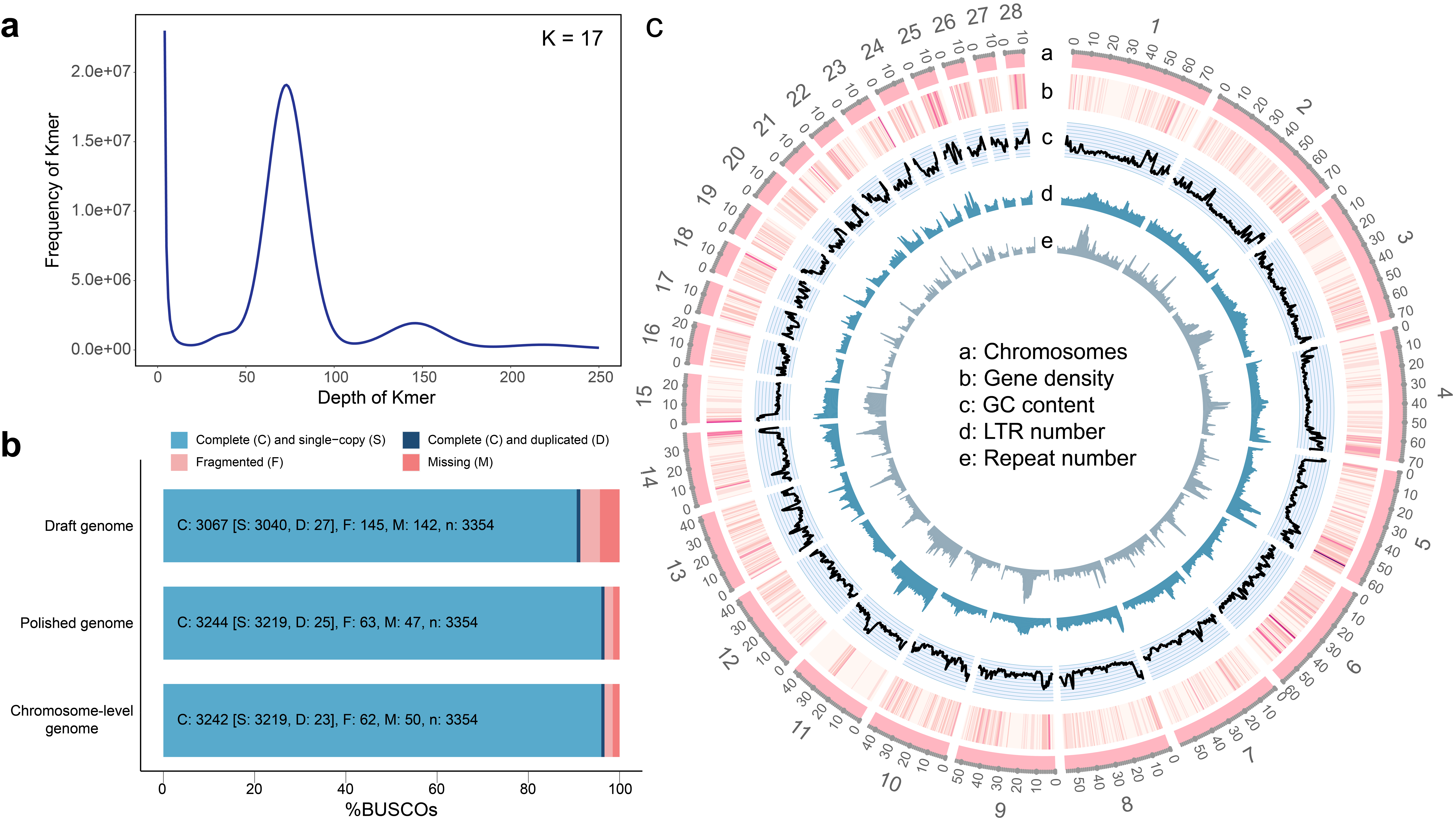
Fig. 1 Genome assembly of the alligator gar. (a) K-mer frequency distribution at k-mer size of 17. K-mer refers to an artificial sequence division of K nucleotides. The peak depth was 73X. The total number of 17-mer present in this subset was 86,674,739,928. (b) BUSCO scores of the draft, polished, and final chromosome-level genome. (c) General view of the alligator gar genome in nonoverlapping 500 kb windows. a: circular map of 28 chromosomes. b: heat map of gene density. The darker the colour, the higher the density. c: GC content. d: Long Terminal Repeat (LTR) number. e: Repeat number.
Key findings
The identification of high-quality genome of the alligator gar linked to evolution and invasiveness is crucial to protect local aquatic ecosystem functions. In this study, we generated a high contiguity, completeness, and accuracy genome assembly of alligator gar at chromosome level using Oxford Nanopore Technology and Hi-C sequencing techniques. A total of 19, 070 protein-coding genes in the genome were functionally annotated. Based on the results of Kyoto Encyclopedia of Genes and Genomes enrichment analysis for remarkable expansion gene families, we found the alligator gar had strong survival and reproductive capacity, which facilitated its rapid spread, acclimation, and propagation in new environments.
Implications
Gene enrichment in antigen processing and presentation, as well as gametogenesis and fertilization, may explain the genetic basis of alligator gar invasiveness. Overall, the findings of this study deepen our understanding of the underlying genomic basis to comprehend the ecology, evolution, and invasiveness of alligator gar, and aid in watershed aquatic ecosystems management for invasive alien species.
Follow the Topic
-
Scientific Data
A peer-reviewed, open-access journal for descriptions of datasets, and research that advances the sharing and reuse of scientific data.

Related Collections
With Collections, you can get published faster and increase your visibility.
Genomics in freshwater and marine science
Publishing Model: Open Access
Deadline: Jul 23, 2026
Computer vision in plant science and agriculture
Publishing Model: Open Access
Deadline: Jul 10, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in