How is the identity of a cell encoded in its genome? And what underlies transcriptomic changes in disease states? SCENIC+ helps researchers provide answers to these questions (and many more … ) by inferring enhancers and gene regulatory networks.
Published in Protocols & Methods


Cells have the potential to undergo many changes during their lifetime. For instance, single-celled organisms are able to change their metabolism in the presence or absence of certain sources of energy in their environment. Another, more complex example is the development of an entire human being from a zygote. The rules on how and when cells will change are encoded into each cell’s genome. The genome is thus complex and not all parts of the genome should be used at the same time. For example, in the human body photoreceptor cells need to be able to have different functions compared to enterocytes in the gut, they should be able to detect light but not absorb nutrients. To account for these functional differences different genes are transcribed in different cell types. This differential control of gene expression is called gene regulation and involves complex gene regulatory networks where transcription factors interact with cis-regulatory elements to control the transcription of target genes. cis-regulatory elements are non-coding parts of the genome that regulate the expression of nearby genes by having binding sites for specific combinations of transcription factors. In the Laboratory of Computational Biology at the VIB-KU Leuven Center for Brain & Disease research we develop computational tools to decode the gene regulatory mechanisms underlying functional differences between different cell types or states.
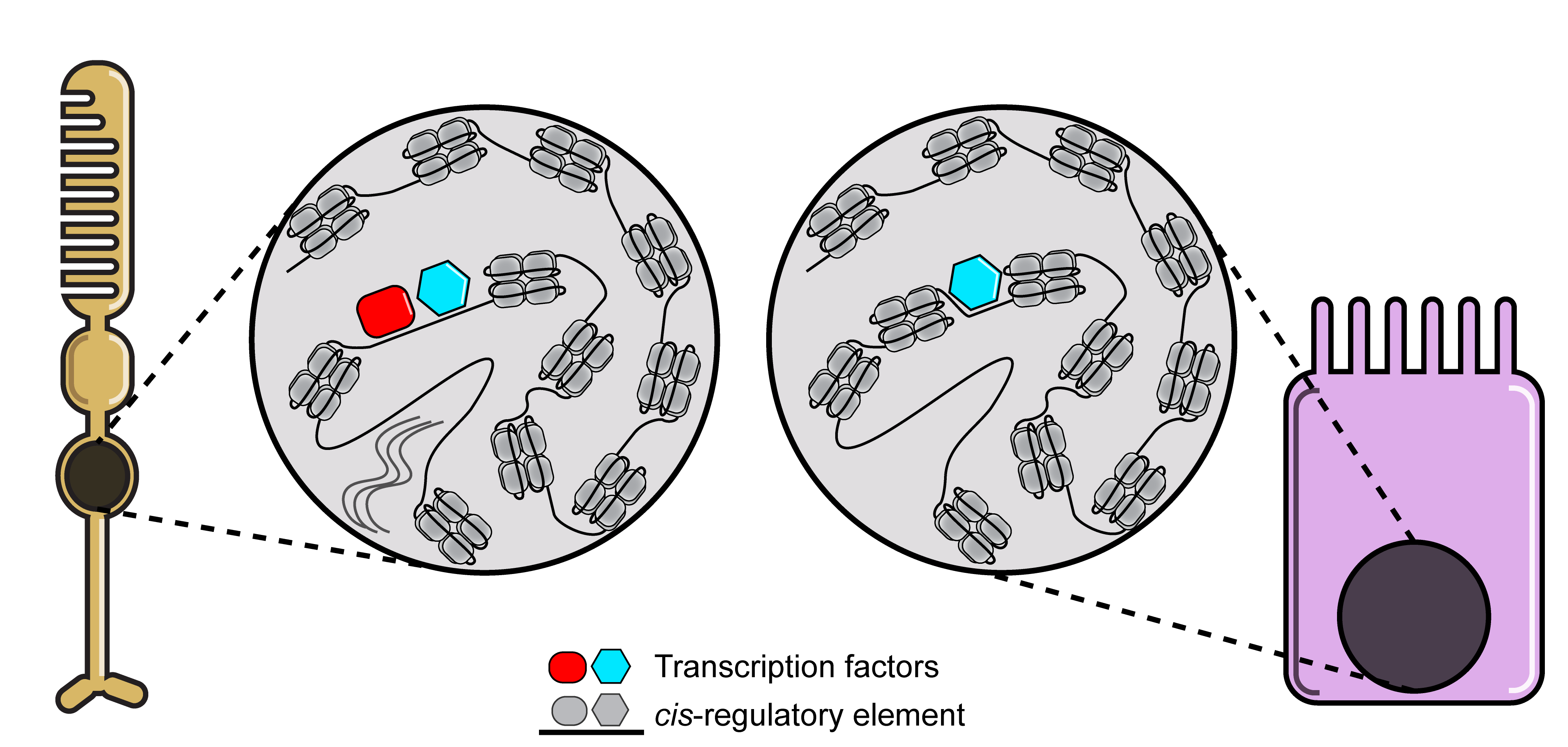
An important thing to keep in mind when decoding gene regulation is that transcription factors do not randomly bind to genomic sequences. Each transcription factor has a preference for certain DNA sequences. Once we know the preference of each transcription factor, we can predict the potential of a specific transcription factor binding based on the sequence of a cis-regulatory element. To learn the preference of each transcription factor, researchers typically use experimental techniques like Chromatin ImmunoPrecipitation followed by sequencing (ChIP-seq) and Consecutive Affinity-Purification Systematic Evolution of Ligands by EXponential (CAP-SELEX) enrichment. These techniques enrich DNA sequences bound by transcription factors, which can then be modeled using Position Weight Matrices (PWMs).
The Laboratory of Computational Biology has been collecting these PWMs from a variety of databases into a big secondary database. The lab also developed a computational algorithm called cisTarget to test for enrichment of PWMs in the DNA sequence of a set of cis-regulatory elements1-54. Using this algorithm, we are able to predict how transcription factors bind to cis-regulatory elements and subsequent expression of target genes based solely on the DNA sequence of these elements. However, this can result in a high number of false positives. To decrease the amount of false positive predictions, the lab developed another computational tool called SCENIC5,6. This tool was developed with the advent of single-cell RNA sequencing (scRNA-seq) and combines motif enrichment with co-expression analysis to limit transcription factor target gene predictions to those predictions where the transcription factor and target gene are co-expressed across individual cells.
For all the tools the lab had developed so far, it was assumed that the cis-regulatory elements of a gene are located somewhere close to the gene’s start/end or in one of its introns. However, the exact locations of these elements were unknown. For this reason, the next challenge became pinpointing the location of these elements, so we can understand which parts of the genome have regulatory importance. To look into this further, we typically use single-cell Assay for Transposase Accessible Chromatin and Sequencing (scATAC-seq) This assay measures which regions of the genome are depleted of nucleosomes (proteins that pack DNA into tightly wound chromatin). Given that transcription factor binding often causes nucleosome depletion, this was a well-suited proxy to finding cis-regulatory elements. To analyze the data generated by this assay a tool called cisTopic was developed in the lab8.
The next step was to use the sequence of the cis-regulatory elements identified through scATAC-seq to predict transcription factor binding. This method results in a smaller amount of DNA sequences having to be analyzed which, in turn, reduces the number of false positives. The reason for this is that the identified elements have a higher chance of being functionally important compared to blindly analyzing the full DNA sequence surrounding a gene. This also allows us to increase the search space around each gene, which is important given that cis-regulatory elements can be located more than 100kb from the start/end of a gene. To further improve the accuracy of predictions, this information can be supplemented with scRNA-seq data. Soon, we showed in the lab that the combination of scATAC-seq with scRNA-seq data analysis can result in accurate predictions of genomic enhancers and high-quality gene regulatory networks9,10.
Finally, we developed SCENIC+ to streamline this process and make it easier for researchers around the world to perform a similar analysis. SCENIC+ is a Python package that contains the tools to analyze scATAC-seq data (pycisTopic) in order to find candidate cis-regulatory elements, analyze the sequence of these elements for the presence of transcription factor binding sites (pycisTarget) and to combine this information with gene expression data to predict gene regulatory networks. Using this new tool the analyses will be faster and it also has many functionalities for visualizing your data. Furthermore we provide methods to predict the functional importance of transcription factors. For instance, SCENIC+ can provide insight into the effect a transcription factor perturbation will have on a certain cell type. This can help researchers with selecting transcription factors to knock down experimentally to induce a certain cell type switch. For example, to change the state of cancer cells. SCENIC+ can also help in providing insight into what the role of a transcription factor is in a differentiation trajectory. This is useful for researchers wishing to understand how one cell type is converted into another during development.
References
-
Potier, D. et al. Mapping gene regulatory networks in drosophila eye development by large-scale transcriptome perturbations and motif inference. Cell Reports 9, 2290–2303 (2014).
-
Herrmann, C., Van de Sande, B., Potier, D. & Aerts, S. I-cistarget: An integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Research 40, (2012).
-
Imrichová, H., Hulselmans, G., Kalender Atak, Z., Potier, D. & Aerts, S. I-cistarget 2015 update: Generalized cis-regulatory enrichment analysis in human, mouse and fly. Nucleic Acids Research 43, (2015).
-
Verfaillie, A., Imrichova, H., Janky, R. & Aerts, S. Iregulon and I‐cisTarget: Reconstructing regulatory networks using motif and track enrichment. Current Protocols in Bioinformatics 52, (2015).
-
Aibar, S. et al. Scenic: Single-cell regulatory network inference and clustering. Nature Methods 14, 1083–1086 (2017).
-
Van de Sande, B. et al. A scalable scenic workflow for single-cell gene regulatory network analysis. Nature Protocols 15, 2247–2276 (2020).
-
Bravo González-Blas, C. et al. CisTopic: Cis-regulatory topic modeling on single-cell ATAC-Seq Data. Nature Methods 16, 397–400 (2019).
-
González-Blas, C. B. et al. Identification of genomic enhancers through spatial integration of single-cell transcriptomics and Epigenomics (2019). doi:10.1101/2019.12.19.882381
-
Janssens, J. et al. Decoding gene regulation in the Fly Brain. Nature 601, 630–636 (2022).
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Jul 28, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in