Human cell transformation by combined lineage conversion and oncogene expression
Published in Cancer
Extensive cancer genome sequencing efforts have greatly contributed to our understanding of human cancer. These studies have reported mutations in over 250 genes in different human tumor types and identified recurrently mutated oncogenes and tumor-suppressor genes that are the likely drivers of transformation and tumorigenesis. However, establishing a causal link between cancer-associated mutations and formation of cancer has been surprisingly difficult, and it is still largely elusive which specific mutations found in human patients lead to tumorigenesis. In our recently published study in Oncogene, we set out to determine what are the factors necessary and sufficient for making cancer from normal human cells. Importantly, our results demonstrate that cellular lineage and differentiation stage are critical factors in transformation by oncogenic drivers.
It is well-established that genetic alterations associated with different cancer types show considerable tissue specificity (1,2); for example, recurrent KRAS mutations are detected in pancreatic and lung tumors but not in breast tumors. However, why particular tissues are vulnerable to specific oncogenic insults while others are not is one of the pertinent questions of modern cancer research. An emerging concept is that tissue specificity in cancer is facilitated by the existing regulatory networks and epigenetic landscape in a given cell, and that cellular identity defines the susceptibility of cells to different growth-promoting signals. Thus, systematic mechanistic studies in the context of correct cellular lineage are warranted for understanding human cancer.
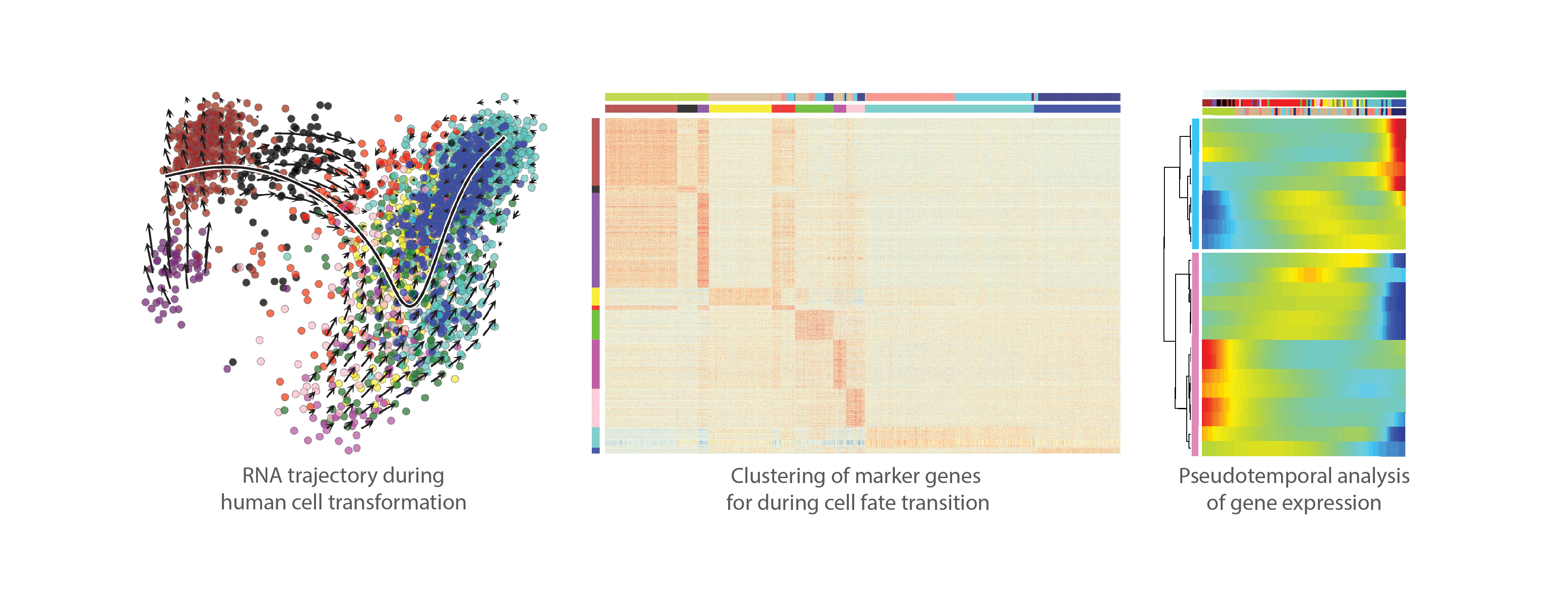
To study the molecular determinants of human tumorigenesis in a defined and systematic manner, we developed a novel cellular transformation assay by combining cell fate conversion and oncogene activation. In this approach, we leveraged the recent advances in the field of cellular transdifferentiation as well as the large body of data regarding cancer-specific mutations catalogued by COSMIC (https://cancer.sanger.ac.uk/cosmic). The discovery by Takahashi and Yamanaka (3) that terminally differentiated cells can be reprogrammed to become pluripotent just by expressing few transcription factors led to development of various protocols for cell fate engineering, including direct transdifferentiation where defined pools of lineage-specific transcription factors are used for directly converting one cell type to another, such as human fibroblasts to induced neurons or hepatocytes (4,5). We reasoned that this provides an appealing platform for studying the effect of oncogenic drivers in the context of cellular lineage, since different immature/progenitor cellular states can be targeted with oncogenes along the course of lineage conversion. Importantly, direct transdifferentiation protocols also skip the pluripotent cellular state that in itself is tumorigenic in mice.
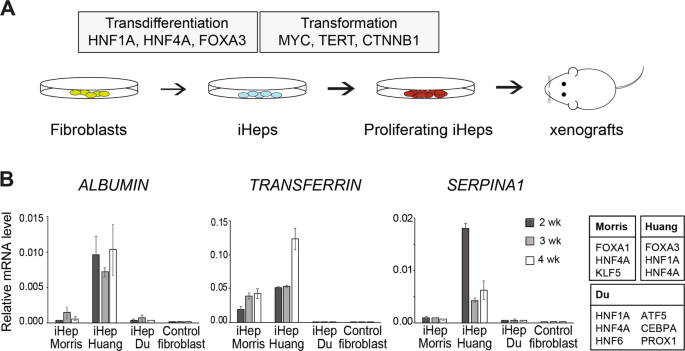
Here, we used three lineage-specific transcription factors (TF) FOXA3, HNF1A, and HNF4A (5) to transdifferentiate human fibroblasts to induced hepatocytes (iHep), and then exposed the iHeps to different combinations of liver cancer-specific oncogenic drivers during cell fate conversion. We identified a pool of three oncogenes, i.e. constitutively active b-catenin (CTNNB1T41A), MYC and TERT that, with or without simultaneous TP53 inactivation using CRISPR/Cas9 system, is able to convert iHeps to a highly proliferative phenotype. Importantly, the transformed iHeps produced tumors in xenograft experiments in nude mice. These results demonstrate that ectopic expression of defined lineage-specific TFs and oncogenes can convert normal human fibroblasts to transformed tumorigenic cells. However, the same cocktail of oncogenes transduced to control fibroblasts or primary human hepatocytes resulted in cellular senescence, suggesting that the cellular lineage and the differentiation state of cells along the lineage defines how susceptible the cells are to transformation.
The tumors derived from transformed iHeps were positive for important liver cancer markers glypican-3 and arginase-1, and their gene expression profile resembles that of the human hepatocellular carcinoma tumors. Moreover, ATAC-seq analysis for open chromatin and NaNoMe-seq for chromatin accessibility and DNA methylation at a single-allele level showed robust changes in chromatin accessibility at the liver and liver cancer marker genes concordant with the observed gene expression changes. Spectral karyotyping revealed that while the parental fibroblasts showed normal diploid karyotype, the transformed iHeps had acquired aneuploidies and chromosomal aberrations typical for hepatocellular carcinoma patients, such as pseudotetraploidy and loss of chromosomes 4 and 13. While genomic instability observed in our model system is typical for almost all human cancers, it can also result in additional cancer-driving mutations besides the oncogenes that were introduced in the experimental system.
Our novel approach that combines cellular reprogramming to oncogene transduction for studying transformation and tumorigenesis has several strengths: first, the use human cells instead of model organisms and the use of oncogenes specific to human cancer types makes it highly relevant for studying human cancer; second, the dynamic assay enables precise molecular control of the cell identity and oncogene expression over the course of the experiment; third, the assay recapitulates the differentiation states between cell identities, creating a cellular state in which cells are susceptible to transformation. Thus, it provides a platform for detailed mechanistic studies for the interplay of cell identity and epigenetic state with oncogenic drivers in any type of human cancer. Wider utilization of the approach could be limited by the availability of the defined factors necessary for generating induced cell types. However, there are computational frameworks such as Mogrify that have been developed for predicting the transcription factors required for reprogramming one cell type to another (6). Moreover, a recent report used a large array of human TFs (Human TFome) for generating a plethora of new induced cell types (7) that, in the future, could be investigated together with the major human oncogenic drivers identified by cancer genetic studies. In conclusion, by studying how normal cells are reprogrammed towards specific cancer types we can ultimately understand how cellular lineage and identity contribute to development of human cancers. This might also reveal novel tissue-specific vulnerabilities that can be used for developing targeted cancer therapies.
- Schneider G, Schmidt-Supprian M, Rad R, Saur D. Tissue-specific tumorigenesis: context matters. Nat Rev Cancer. 2017;17:239-253.
- Haigis KM, Cichowski K, Elledge SJ. Tissue-specificity in cancer: The rule, not the exception. Science. 2019;363:1150-1151.
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-676.
- Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, et al. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476:220-223.
- Huang P, Zhang L, Gao Y, He Z, Yao D, Wu Z, et al. Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell Stem Cell. 2014;14:370-384.
- Rackham OJ, Firas J, Fang H, Oates ME, Holmes ML, Knaupp AS, et al. A predictive computational framework for direct reprogramming between human cell types. Nat Genet. 2016;48:331-335.
- Ng AHM, Khoshakhlagh P, Rojo Arias JE, Pasquini G, Wang K, et al. A comprehensive library of human transcription factors for cell fate engineering. Nat Biotechnol. 2021;39:510-519.
Follow the Topic
-
Oncogene
This journal aims to make substantial advances in our knowledge of processes that contribute to cancer by publishing outstanding research.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in