Human gut microbiome and fecal metabolome: a detailed description across three generations reveals the uniqueness of infants vs their elders
Published in Chemistry, Microbiology, and Paediatrics, Reproductive Medicine & Geriatrics
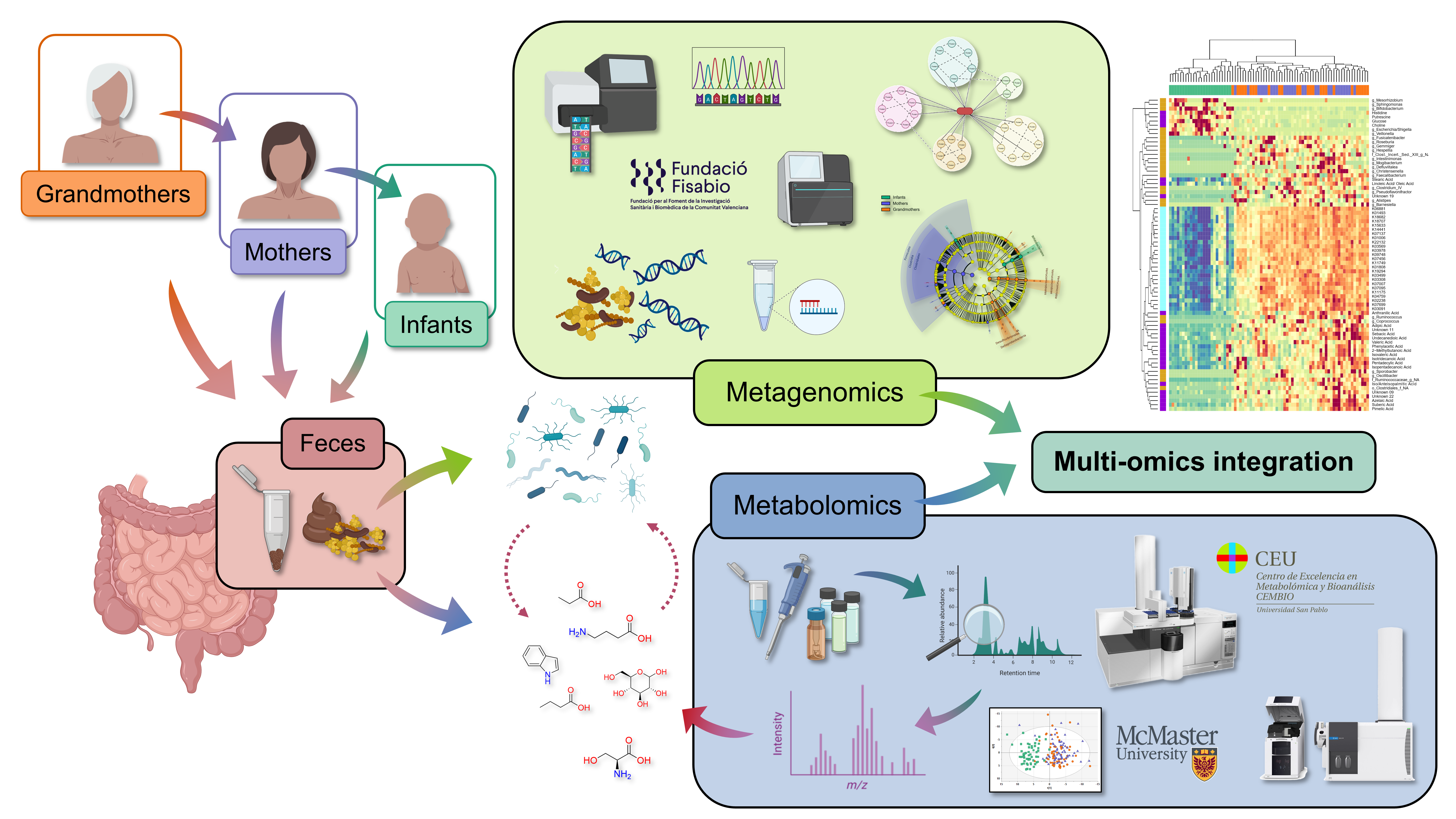

Introduction and Study Design
The human gut microbiome constitutes a complex ecosystem that plays a fundamental role in human health and disease. It establishes and matures during infancy, and dysregulation at this stage may lead to pathologies later in life. Although the importance of the gut microbiome is widely recognized, there is still much to learn about its early development and how it influences human health over the lifespan.
This study explores the intergenerational comparison of the gut microbiome and fecal metabolome in infants, their respective mothers, and grandmothers. We recruited 200 participants across these three generations in two main Hospitals and several Healthcare Centers in Madrid, Spain (Figure 1). This was achieved thanks to the close collaboration with clinicians Dr. M. Dolores Ibáñez-Sandín, Dr Raphaëlle Bazire, and Dr. Paula Cabrera-Freitag. Exclusion criteria included recent antibiotic intake and severe diseases, ensuring a relatively healthy study population. It is worth highlighting that the recruitment process spanned two years due to the challenge of finding families with all three generations willing to participate.

Metabolomics analyses
After this recruitment process, we performed the analysis of the fecal samples in gas chromatography coupled to quadrupole time-of-flight mass spectrometry (GC-QTOF-MS). Large-scale analyses in this technique are uncommon, since the samples need to be derivatized with complex and time-consuming protocols. To overcome this issue, we applied a short derivatization process that significantly reduced sample preparation times. Additionally, we applied a batch strategy with a common quality control sample, which was later used to normalize the data from the different batches. The success of this normalization strategy was proven by several data quality parameters and enhanced the reliability of the results, ensuring data accuracy and consistency. This is one of the main strengths of this article and provides a useful blueprint for future studies using this technique.
After the quality of the analysis was checked, the differences between the groups (infants, mothers, and grandmothers) considering their family link were obtained. The first result we observed was that infants greatly differed from their elders in the fecal metabolome, as they separated very clearly in the multivariate models and presented a high number of significantly altered metabolites. These remarkable results made us believe we had something very valuable in our hands.
Infants greatly differed from their elders in the fecal metabolome, as they separated very clearly in the multivariate models and presented a high number of significantly altered metabolites. These remarkable results made us believe we had something very valuable in our hands.
However, we were missing some important metabolites of the host-gut microbiota interaction that we had not been able to detect with GC-QTOF-MS. This proved the perfect opportunity for Tomás, the lead author of the paper, to carry out his international PhD research stay, to train in a novel metabolomics technique –capillary electrophoresis coupled to mass spectrometry with an innovative application of consisting of multisegment injection (MSI-CE-MS)– and to establish an international collaboration with Dr. Philip Britz-McKibbin and Dr. Meera Shanmuganathan from McMaster University in Canada.
This collaboration proved fruitful, as the analysis was successful and provided the missing metabolites –e.g., butyric/isobutyric acid, γ-aminobutyric acid (GABA), and glucose– that confirmed the findings from GC and completed the metabolomics puzzle. Taken together, the outcomes from both metabolomics analyses (GC-QTOF-MS and MSI-CE-MS) confirmed the previously seen result: infants greatly differed from their elders in their fecal metabolome.
Among the significant metabolite classes, amino acids, Krebs cycle intermediates, indoles, and mainly fatty acids (including short- and branched-chain fatty acids, SCFA and BCFA) were found. Specifically, while the levels of saturated fatty acids (i.e., palmitic and stearic acids), monounsaturated fatty acids (MUFA; e.g., oleic acid) and linoleic acid were higher in adults, the opposite was observed for polyunsaturated fatty acids (PUFA; e.g., arachidonic and docosahexaenoic acids). This could potentially be linked to changes in dietary habits and metabolic processes related to age.
Regarding SCFAs, infants displayed higher levels of acetic acid and lower amounts of the other SCFAs, namely propionic, (iso)butyric, caproic, and (iso)valeric acids. Similarly, all BCFAs were significantly increased with age.
Metagenomics and multi-omics integration
Having the full picture from the metabolomics perspective, we decided it was worth it to put in the extra effort and perform the integration with the metagenomics data from a subset of these samples, to delve into the functional and taxonomic diversity of the gut microbiome. This data was obtained in collaboration with the FISABIO research center in Valencia thanks to Dr. Carles Úbeda and Dr. M. Pilar Francino.
First, we compared the bacterial communities between the age groups in terms of taxonomy using 16S rRNA gene sequencing data. We found that, compared with adults, Infants have a less diverse microbiota, which is associated with shifts in bacterial populations. Notably, infants exhibited a higher proportion of Actinobacteria and Proteobacteria phyla, while adults had a predominant presence of Firmicutes phylum. Moreover, significant disparities were found in the relative abundance of genera like Bifidobacterium, Escherichia/Shigella, and Veillonella, much higher in infants, compared to Faecalibacterium, Blautia, and Roseburia, much higher in adults.
Correlations between these microbial abundances and metabolites such as GABA and SCFAs were statistically significant, underlining the metabolic implications of these differences in gut microbiota (Figure 2).

Furthermore, the application of shotgun sequencing allowed a deeper understanding of the taxonomic composition in the groups, showing that a few species, including Bifidobacterium bifidum, B. breve, B. longum, Escherichia coli, and Faecalibacterium prausnitzii, accounted for 40% of the gut microbiota in infants. The shotgun sequencing data also provided information about the functional capacity of the gut microbiota, revealing that many pathways related to energy and carbohydrate metabolisms were more abundant in infants. On the other hand, functions related to polyamines signalling and BCFAs metabolism were increased with age. From these findings we conclude that not only is the microbiota of infants different, but it also behaves differently and is accountable for the changes that we see between age groups.
Finally, we integrated all these results together –fecal microbiota genera, function, and metabolites. This was thanks to the hard work of Lola Alonso and Dr. Isabel Adoración Martín-Antoniano. Although each omics technique showed good discrimination parameters by itself, it was only this integration that allowed us to draw an almost perfect multi-omics fingerprint of the microbiota of Infants. This process was complemented by a literature review and by the careful curation of the data, thanks to which we built detailed pathways –including the bacterial enzymes involved as well as the actual metabolites. These significantly strengthened our biological interpretation and provided compelling evidence of the interrelated processes.
Conclusion and significance of the study
In conclusion, this study provides important insights into the intergenerational differences in the gut microbiome and metabolome, shedding light on the early development of these microbial communities. The findings highlight the importance of further research to understand the complex interactions between the gut microbiota, metabolites, and host health, and emphasize the need for longitudinal studies that follow participants over time and assess long-term health outcomes.
This study provides important insights into the intergenerational differences in the gut microbiome and metabolome, shedding light on the early development of these microbial communities.
In general, we provide extensive open-access data from each of the omics techniques in our supplementary datasets, source data, and in the repositories. In addition, we have taken great care to provide aesthetically pleasing and high-quality figures, graphs, and tables, with a consistent colorblind-friendly palette to easily compare the findings of the different omics techniques. This proved to be no easy task, especially for the graphs from the mixOmics results, which had to be painstakingly recolored and formatted.
Overall, this work is the result of years of effort and the hard work of many people, and we believe it constitutes a significant contribution to the field and a valuable resource for future studies linking the aging process to the microbiota and its associated metabolites.
Authors of the paper:
Social Networks and Webpages:
-
X (formerly Twitter):
- CEMBIO: @metabolomicaceu
- Allerbiomics: @allerbiomics
- Britz Group McMaster: @britzgroup
-
Webpages:
- CEMBIO: https://cembio.uspceu.es/
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in