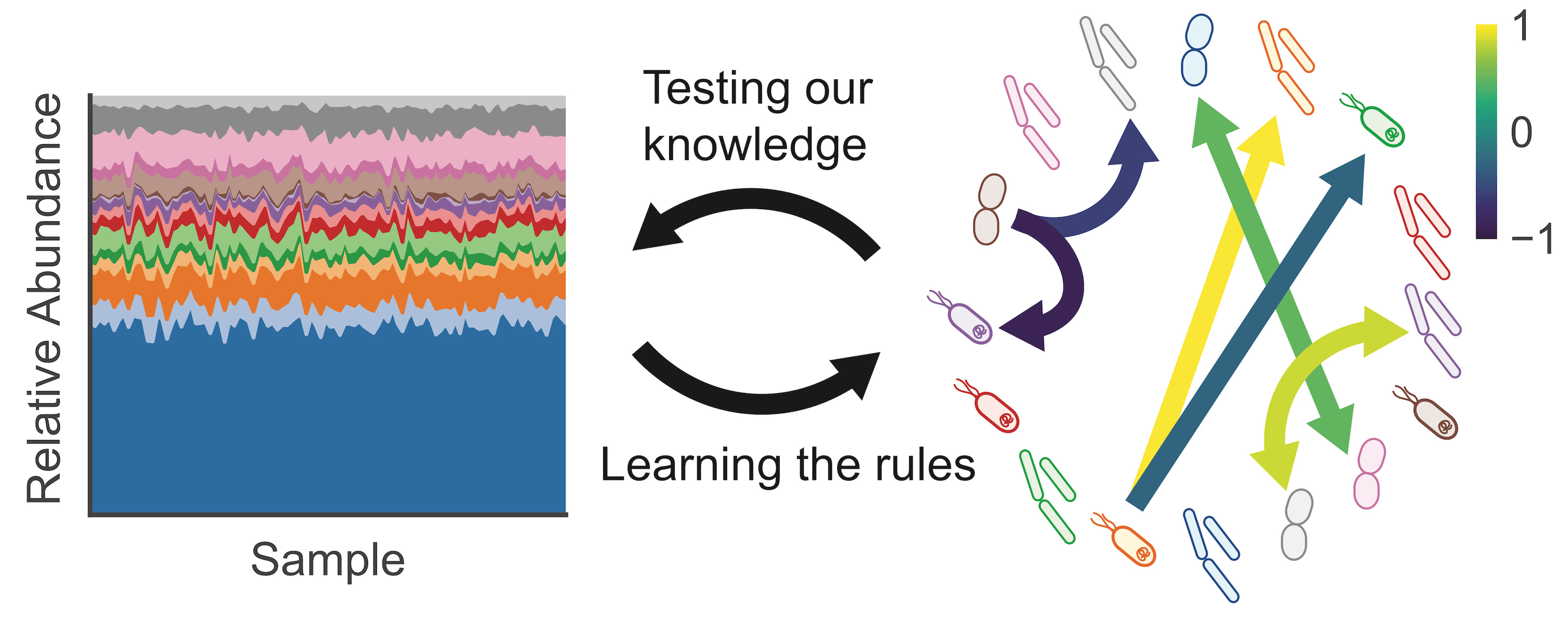
Inferring microbial interactions from relative abundance: not as easy as you would think
Published in Microbiology
Explore the Research

Use and abuse of correlation analyses in microbial ecology - The ISME Journal
The ISME Journal - Use and abuse of correlation analyses in microbial ecology
Our ability to characterize microbial communities in their native habitats with 16S amplicon sequencing and shotgun metagenomics has revolutionized the study of microbial ecology. These techniques have given us access to a vast amount of information about microbial diversity in the ocean, the soil, and our bodies. Information that has made it possible to explore relationships between microbial abundance, community structure, and ecosystem properties. Human gut microbiome studies have used sequencing data to establish links between diet and community composition, identify associations between gut dysbiosis and disease, and provide a rational for the therapeutic application of fecal microbiome transplants, among other important advances [1].
My entry into the field came prior to my graduate studies, when I joined Adam Arkin’s lab at UC Berkeley as a post-undergraduate research associate. It was there that I had the privilege of working with an amazing group of colleagues on a project to experimentally characterize species-species interactions within a model community of twelve human gut bacterial isolates. This experience opened my eyes to the world of microbial ecology and led to a publication where we demonstrated that dynamics in our synthetic consortia could be predicted with relatively high accuracy using a bottom up approach [2]. However, even in our relatively simplified model system, where the growth conditions were held constant, there was no spatial heterogeneity or dispersal, and high-resolution time series measurements of absolute abundances for each species were obtained, some interactions remained elusive. A few of the organisms had a significant impact on multi-species community composition that could not be predicted from pairwise associations. Similarly, others have found it challenging to accurately model microbial interactions in simplified microcosms - this is not a trivial endeavor [3,4]. As such, it should not come as a surprise that in more complex environments - like the human gut or the soil - interaction characterization is much more difficult.
Complex nonlinear dynamics, compositionality of the data, environmental heterogeneity, latent variables, and batch effects are all factors that can hinder the utility of correlation analyses and misinform hypotheses about interactions derived from fluctuations in relative abundances in real-world environments. Nonetheless, there has been a significant amount of progress made in addressing many of these challenges. With careful consideration of the various pitfalls and limitations, and a healthy dose of skepticism, sequencing surveys can be a useful means of generating hypotheses about biotic interactions.
In our recent Perspective article in the ISME Journal, we provide a discussion of the challenges associated with interpreting microbial correlation networks and highlight some of the efforts made by the bioinformatics community to improve interaction inferences. Integrating other types of data into correlation analyses, like measures of potentially confounding environmental variables, accurate noise and bias estimates, absolute biomass, the ordering of events in space or time, multi-omic measurements, and mechanistic constraints can greatly improve inferences. However, as we state in our Perspective, “bioinformatic approaches only generate hypotheses. In order for these inferred interactions to be accepted as truth, the hard work of experimental validation is required.”
Article link: rdcu.be/bH0Ue
References:
- Parient N. (2019). Milestones in human microbiota research. Nature. https://www.nature.com/immersive/d42859-019-00041-z/index.html
- Venturelli OS, Carr AC, Fisher G, Hsu RH, Lau R, Bowen BP, et al. (2018). Deciphering microbial interactions in synthetic human gut microbiome communities. Molecular Systems Biology.
- Friedman J, Higgins LM, Gore J. (2016). Community structure follows simple assembly rules in microbial microcosms. Nature Ecology & Evolution.
- Hart SFM, Mi H, Green R, Xie L, Pineda JMB, Momeni B, et al. (2019). Uncovering and resolving challenges of quantitative modeling in a simplified community of interacting cells. PLOS Biology.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in