
RAS mutations prevalent in high-risk leukemia have been linked to relapse and chemotherapy resistance. Efforts to directly target RAS proteins have been largely unsuccessful until recently with the approval of sotorasib, a KRAS G12C specific inhibitor. However, G12C mutations are very rare in acute lymphoblastic and myeloid leukemias (ALL and AML). Since RAS-mediated transformation is dependent on signaling through the RAS-related C3 botulinum toxin substrate (RAC) small GTPase, we hypothesized that targeting RAC may be an effective therapeutic approach in RAS mutated tumors. In our article entitled “Validation of a small molecule inhibitor of PDE6D-RAS interaction with favorable anti-leukemic effects” we report the identification of a set of compounds that demonstrated dose-dependent RAC inhibition, arrest of proliferation, and induced apoptosis in human leukemic cell lines. However, none of them showed inhibition of the RAC1-TIAM1 protein-protein interaction meaning that there was no direct inhibition of RAC by these compounds. Given that these small molecules had very promising anti-leukemic activity we embarked on a journey to unravel the mechanisms of action of our most promising compound DW0254. This molecule showed reasonable, but not stellar, lipophilicity and solubility while having very potent biological activity.
Firstly, we developed a photoprobe consisting of DW0254 covalently linked to a minimalist terminal propargyl-diazirine photocrosslinker, referred to as PAL. The PAL-linked probe possessed antiproliferation properties consistent with those of its parent compound, demonstrating that the photoprobe retained activity and was cell-permeable. Using cellular photoaffinity labeling methods combined with label-free quantitative mass spectrometry analysis (PAL-MS) we identified retinal rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiester 6 subunit delta (PDE6D) as a target hit. PDE6D acts as a carrier for prenylated cargo including RAS. These prenylated proteins remain soluble in the aqueous environment of the cytosol only with the help of chaperones. PDE6D specifically sequesters GTP-RAS farnesyl residue in its hydrophobic pocket and helps in transporting it to the plasma membrane where it activates downstream signaling pathways.
Next, we developed a saturating CRISPR mutagenesis screen to identify mutations in PDE6D regions that lead to compound resistance, hoping these would shed some light into the binding mode of DW0254 on PDE6D. We were able to establish that specific mutations in PDE6D’s hydrophobic pocket (R48del V49del) led to compound resistance. Interestingly, DW0254 resistant cells were not resistant to Deltarasin, a known inhibitor of PDE6D that binds the same pocket. We followed up with crystallographic studies where the cocrystal structure of DW0254 with recombinant PDE6D showed the small molecule bound inside the hydrophobic pocket. DW0254 undergoes hydrogen bonding interactions with glutamine Q88, tyrosine Y149, and arginine R61, the latter interaction being water-mediated. Deltarasin occupies the same pocket and forms hydrogen bonds with the same residues R61 and Y149, but also with cysteine C56. By superimposing the binding poses of DW0254 and Deltarasin we were able to contextualize the crystallographic binding modes with the saturating mutagenesis screen results, highlighting that V49 defines the shape of the pocket (light grey area, Figure 1), and establishes hydrophobic contacts only with DW0254 (cyan) but not Deltarasin (orange).
 Figure 1: Experimental binding mode of DW-0254 (cyan) in wild-type PDE6D superposed with Deltarasin (orange) binding mode. Several binding site residues, including V49, are shown for reference.
Figure 1: Experimental binding mode of DW-0254 (cyan) in wild-type PDE6D superposed with Deltarasin (orange) binding mode. Several binding site residues, including V49, are shown for reference.
Since the biological effects of RAS proteins are exerted from the plasma membrane through the activation of kinase pathways including PI3K/AKT and MAPK/ERK, we then analyzed the level of PDE6D-RAS complexes, RAS localization, and downstream pathways activation before and after DW0254 treatment. By co-immunoprecipitating PDE6D with its binding proteins, we observed an almost complete loss of PDE6D-RAS interactions upon treatment, coupled with a decrease in PDE6D-ADP-ribosylation factor-like protein 2 (ARL2) complexes, essential for cargo displacement from PDE6D. With the help of fluorescent-tagged mutant RAS proteins, we also observed loss of RAS membrane localization by real-time imaging upon DW0254 treatment. The story becomes a little more complex when it comes to downstream pathway inhibition. Sensitivity to DW0254 treatment did not correlate with specific RAS pathway mutations and we saw the same range of inhibition of AKT and ERK phosphorylation in both sensitive and resistant cell lines, meaning that the levels of inhibition of RAS downstream pathways do not correlate with compound sensitivity. However, using “The Genomics of Drug Sensitivity in Cancer Project” platform we found a positive correlation between the response of T-ALL cell lines to most AKT inhibitors and to DW0254 (Table 1). This suggests that PDE6D inhibition effectively triggers a downstream anti-leukemic response in AKT-dependent cell lines.
Table 1: Response to AKT inhibitors in T-ALL cell lines from “The Genomics of Drug Sensitivity in Cancer Project” database compared to sensitivity to DW0254. Pearson’s correlation coefficient (r) and the p-value for the correlation coefficient between response to DW0254 and each AKT inhibitor are shown on the right extremity.
Finally, to go full circle and establish a biochemical link between the initially observed GTP-RAC inhibition and RAS pathway inhibition we examined if RAC activation is affected by the direct inhibition of PI3K (LY294002 inhibitor) or MEK (U0126 inhibitor) in a T-ALL model cell line. A consistent decrease in GTP-RAC levels was observed only upon PI3K/AKT pathway inhibition (Figure 2).
 Figure 2: Levels of AKT, ERK, and RAC activation upon treatment with increasing doses of DW0254, LY294002, or U0126.
Figure 2: Levels of AKT, ERK, and RAC activation upon treatment with increasing doses of DW0254, LY294002, or U0126.
Since our initial in vivo pharmacokinetics (PKs) assays demonstrated low solubility and rapid plasma clearance of DW0254, which meant direct in vivo administration by IP or gavage could not be performed, we exploited a drug delivery strategy using Alzet osmotic pumps. Using a luciferase-expressing T-ALL cell line xenograft model to test drug efficiency in vivo over 2 weeks we observed decreased tumor progression in DW0254 treated mice when compared to controls (Figure 3). Notably, control mice exhibited luminescent signal in the abdominal area corresponding to the spleen while DW0254-treated mice demonstrated no observable signal and had a lower percentage of blasts in the peripheral blood (1.8% when compared to 4.2% in the controls).
 Figure 3: Bioluminescent images of day 24 post-transplant of mice treated with DW0254 or Vehicle pumps from day 7 to day 21 and magnified abdominal view.
Figure 3: Bioluminescent images of day 24 post-transplant of mice treated with DW0254 or Vehicle pumps from day 7 to day 21 and magnified abdominal view.
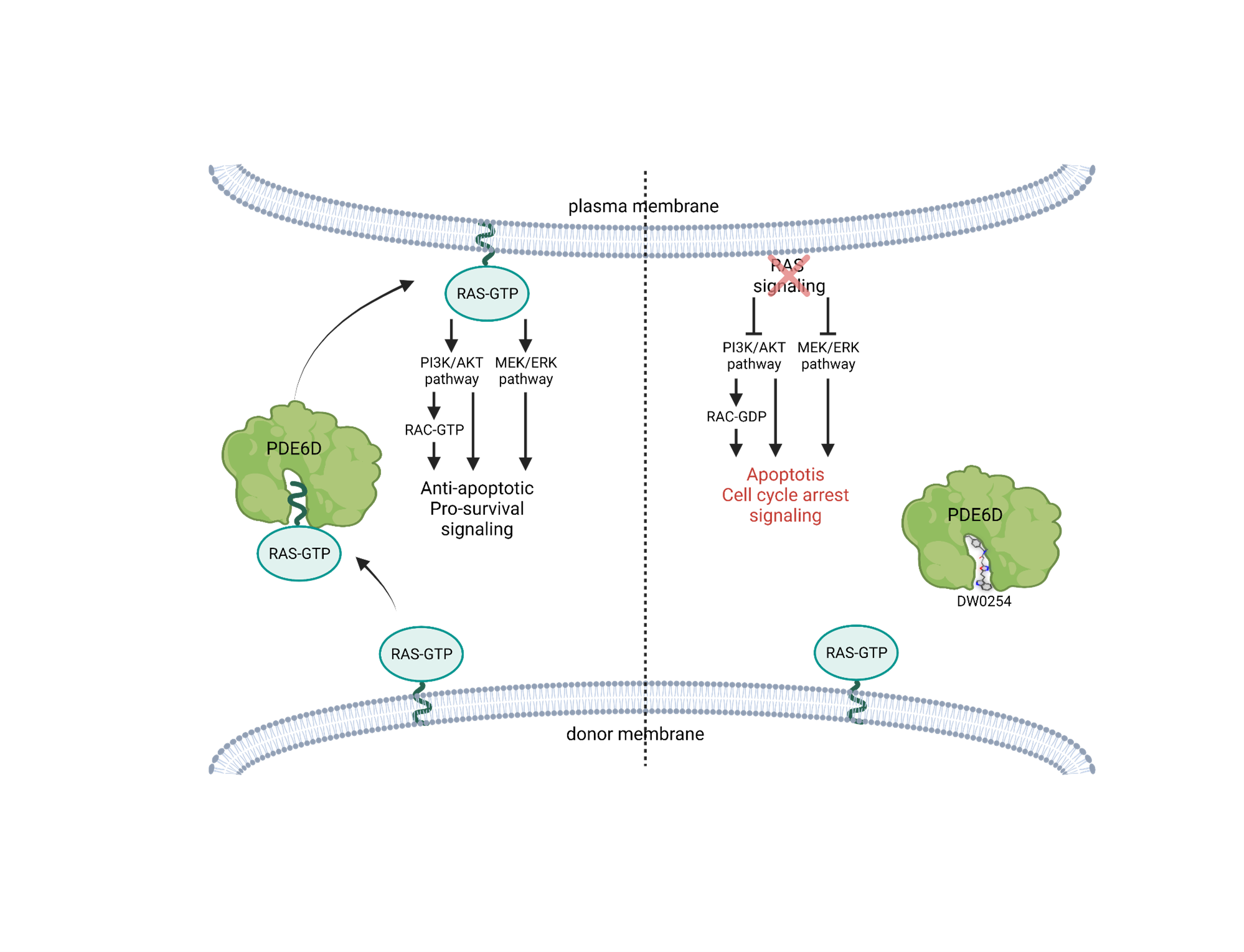
In conclusion, we have validated the RAS chaperone PDE6D as a novel molecular target for aggressive leukemias. The binding of our molecule DW0254 to PDE6D results in the delocalization of RAS from the membrane and consequent inhibition of major leukemic pro-survival pathways including MAPK/ERK, PI3K/AKT, and downstream RAC activation (Figure 4). Sensitivity to DW0254 correlates with sensitivity to AKT inhibition but not to specific RAS pathway mutations. Lastly, were able also to recapitulate the antileukemic results observed in vitro, in a leukemia xenograft model showing decreased tumor progression in DW0254 treated mice. Whether or not the direct inhibition of RAC is an effective therapeutic approach in RAS mutant and/or AKT-dependent leukemias remains an open question.
 Figure 4: Proposed mechanism of action of DW0254 molecule upon binding to PDE6D -created with BioRender.com.
Figure 4: Proposed mechanism of action of DW0254 molecule upon binding to PDE6D -created with BioRender.com.
Follow the Topic
-
Blood Cancer Journal
This journal seeks to publish articles of the highest quality related to hematologic malignancies and related disorders.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in