Irreproducibility in phylogenetic inference
Published in Ecology & Evolution

Over the years, the use of phylogenetic trees has permeated the study of all of evolution and biology in general. Nowadays, state-of-the art phylogenetic inference methods, especially ones designed to analyze large genome-scale data sets, use hill-climbing tree heuristic search algorithms to explore the space of all possible trees. That means that different runs of the same data set may yield different results, especially if the parameters we use (e.g. random starting seed number) differ. However, if we stick to exactly the same parameters including sequence alignment, program, substitution model, random starting seed number, number of tree search, and log-likelihood epsilon value, can we reproduce phylogenetic trees across multiple replicates?
In a summer morning in 2019, I went to the Rokas Lab at Vanderbilt University as usual, in which I had wonderful life and fruitful research achievements, and submitted several jobs to run phylogenetic analyses using the IQ-TREE software on Vanderbilt’s supercomputer, known as ACCRE. When all my analyses were finished, I found that I had accidentally ran some analyses twice (both had the identical parameter settings including random starting seed number). But to my amazement, I realized that these identical runs produced phylogenetic trees that were topologically different from each other. How could this happen? And why did it happen?
To figure the answers to these questions, I decided to rerun the analyses again, and found irreproducible gene trees were still observed across replicates, suggesting that what I was observing, was irreproducibility that is the inability to obtain the same result when the exact same analysis is performed.
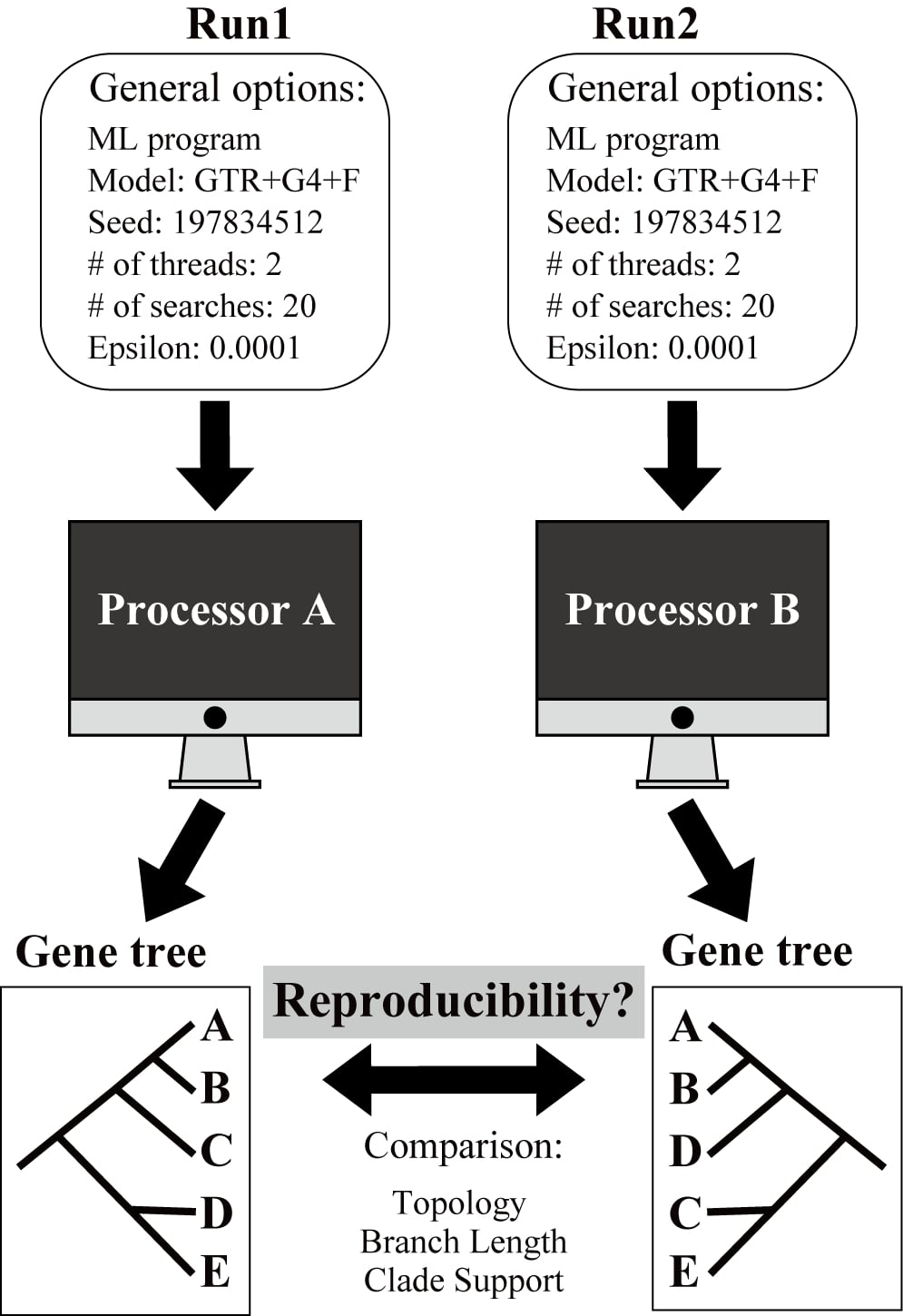
As I finished my postdoctoral training and estimated the Shen Lab at Zhejiang University in December 2019, I started to examine the nature of the irreproducibility of maximum likelihood (ML) phylogenetic trees and the extent to which it affects the analyses of genome-scale or phylogenomic datasets. I collected 19,414 gene alignments from 15 animal, plant, and fungal phylogenomic datasets, for which we included the largest (as of February 2020) available phylogenomic datasets in animals (Marine fishes: 1,001 genes and 120 taxa from Alfaro et al. 1), plants (Green plants: 410 genes and 1,178 taxa from 1KP Initiative 2), and fungi (Budding yeasts: 2,408 genes and 343 taxa from Shen et al. 3). For each gene alignment, I conducted two replicate runs (Run1 and Run2) using identical settings, including substitution model, random starting seed number, number of threads of execution (2), the number of independent tree searches (20), log-likelihood epsilon value (0.0001), and ML program (IQ-TREE or RAxML-NG).
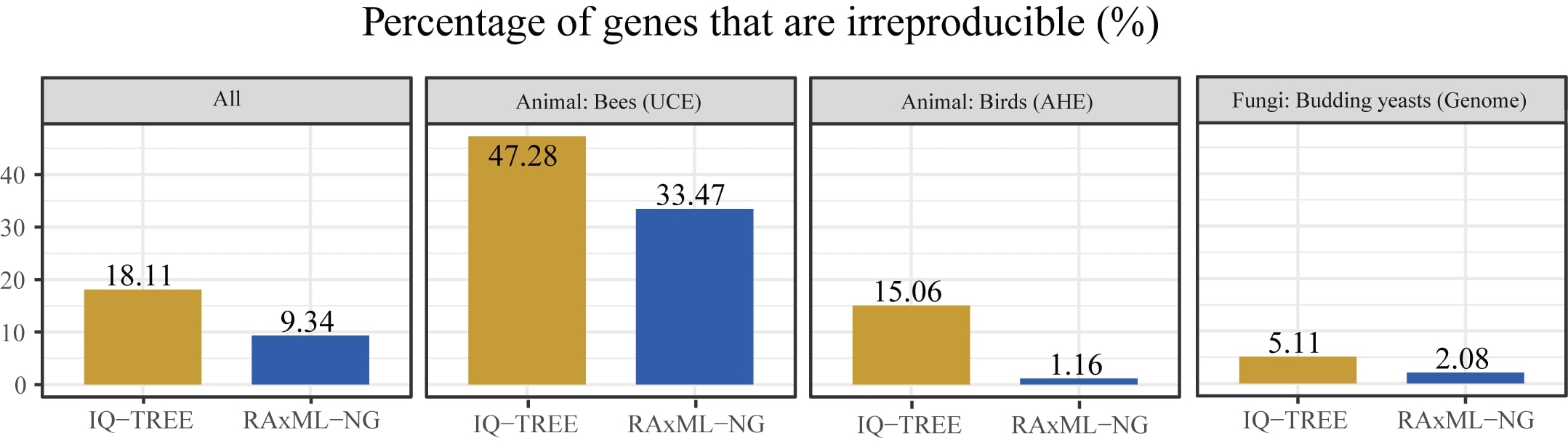
As a result, we found that 3,515 / 19,414 (18.11%) IQ-TREE-inferred gene trees and 1,813 / 19,414 (9.34%) RAxML-NG-inferred gene trees were topologically irreproducible. In addition, we found that irreproducibility was much higher (25.2%) in the eight datasets constructed using ultra-conserved element (UCE) capture 4, Anchored Hybrid Enriched (AHE) capture 5, or conserved exon capture 6 approaches, which tend to generate short alignments of highly conserved gene regions, than in the remaining seven studies (8.5%). Interestingly, we found that using 3 or more threads contributes to 2-fold increase in irreproducibility in IQ-TREE but not in RAxML-NG.
Since the phylogenomic data matrices are typically analyzed using concatenation- and coalescent-based approaches, which have become standard for phylogenomic inference of species phylogenies, we assessed their reproducibility. We found that coalescent-based species phylogeny inference, which relies on separately estimated ML gene trees, is more likely to be irreproducible than concatenation-based ML species phylogeny species, which relies on the supermatrix of gene alignments.
By comparing the characteristics of genes that yielded topologically irreproducible phylogenies (3,515 in IQ-TREE and 1,813 in RAxML-NG) to those that yielded topologically reproducible phylogenies (15,899 in IQ-TREE and 17,601 in RAxML-NG), we found that genes with lower phylogenetic informativeness (e.g., low percentage of parsimony-informative sites in gene alignment, short alignment length, and low branch support values), processor type, and multithreading contribute to the observed irreproducibility, with the effects of multithreading being program-specific.
How can we increase the reproducibility of phylogenetic inference? Given that the benefits need to be weighed against the practical difficulty of dealing with the hundreds or thousands of gene alignments present in current phylogenomic datasets. Moving forward, a more practical alternative may be the releasing of the log file of each analysis, which contain a record of the values of all these key parameters (e.g., alignment, program name, number of tree searches, substitution model, type of processor, number of threads, and random starting seed).
The paper published in Nature Communications can be found here.
References
- Alfaro, M. E. et al. Explosive diversification of marine fishes at the Cretaceous-Palaeogene boundary. Nat. Ecol. Evol. 2, 688–696 (2018).
- One Thousand Plant Transcriptomes Initiative. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685 (2019).
- Shen, X.-X. et al. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 175, 1533–1545 (2018).
- Faircloth, B. C. et al. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst. Biol. 61, 717–726 (2012).
- Lemmon, A. R., Emme, S. A. & Lemmon, E. M. Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst. Biol. 61, 727–744 (2012).
- Bragg, J. G., Potter, S., Bi, K. & Moritz, C. Exon capture phylogenomics: efficacy across scales of divergence. Mol. Ecol. Resour. 16, 1059–1068 (2016).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in