Keeping the balance is important. Also for viruses.
Published in Microbiology and Cell & Molecular Biology
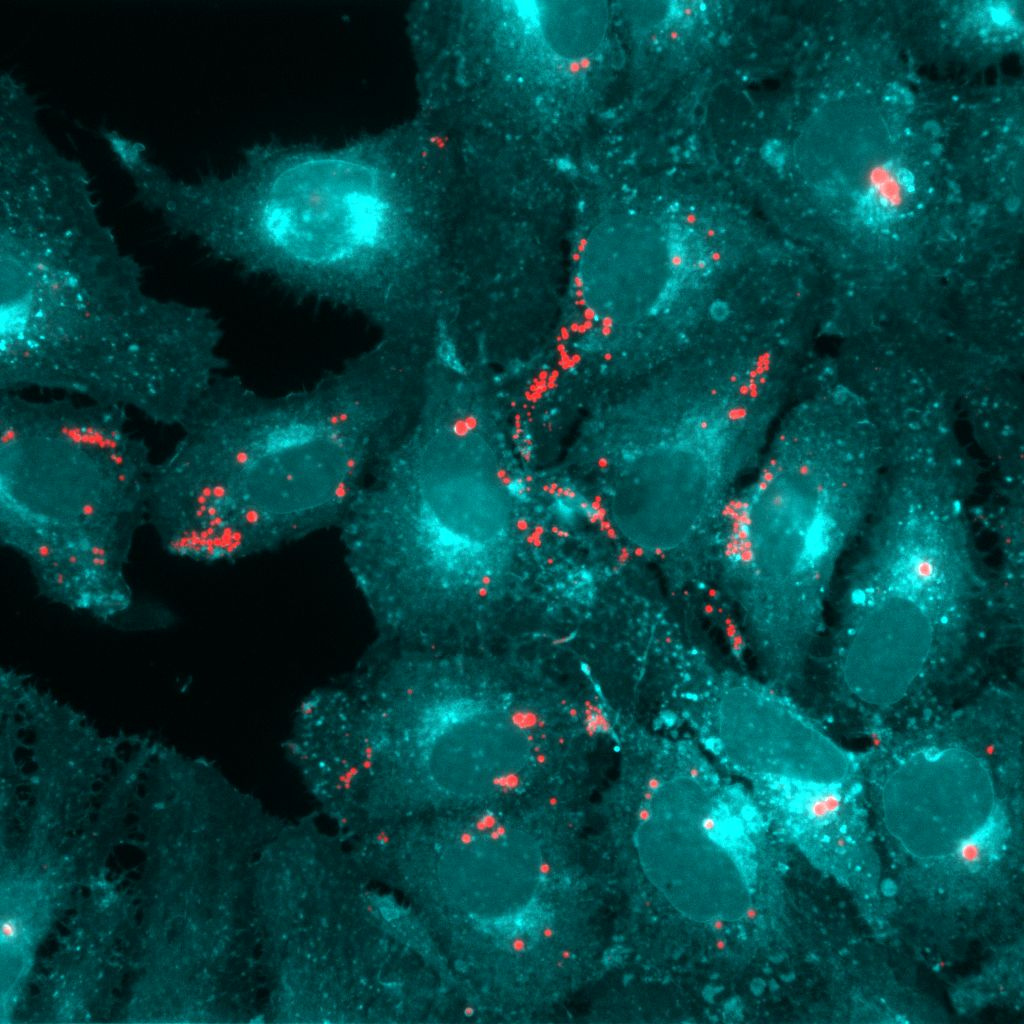
Suppressing virus infection or replication can be a tough nut to crack. You´ve developed an antiviral drug? Let me change my amino acid sequence to overcome this challenge so I can multiply in peace. You have a vaccine? This season, I will definitely mutate the respective epitope. Of course, there are potent functional drugs available for antiviral treatment, but the risk of antiviral drug resistance should not be underestimated. Good thing that viruses are inevitably dependent on one thing: The host. Creating a hostile environment for the virus by targeting essential (cellular) host factors is another approach for antiviral therapy. Remember the roommate that was always letting the dishes pile up in the sink and leaving his stuff everywhere? Imagine creating such an uncomfortable environment for the virus.
Our research group, led by Prof. Dr. Eva Herker, studies the interplay between human pathogenic flaviviruses such as arthropod-borne Zika (ZIKV), West Nile (WNV), tick-borne encephalitis virus (TBEV), and dengue virus (DENV), as well as hepatitis C virus (HCV) and host cell lipid metabolic processes. Various publications have shown that these enveloped positive-strand RNA viruses are highly dependent on a variety of cellular lipids. Thus, investigating the role of lipid-synthesizing enzymes somehow suggested itself.
As part of the LOEWE center DRUID (Novel Drug Targets against Poverty-related and Neglected Tropical Infectious Diseases, [https://www.loewe-druid.de/en/]) we were eager to identify potential antiviral targets for orthoflavivirus infections and started by depleting key enzymes in different cellular lipid pathways using RNAi. As initial cell culture model, we used human hepatoma cells (Huh7), as they are permissive to all the viruses we study (DENV, WNV, TBEV, and ZIKV). Soon, it became evident that neutral lipid-synthesizing enzymes in particular seem to play a role in orthoflavivirus replication. Within the cell, neutral lipids (triglycerides and cholesterol ester) are stored in lipid droplets, organelles that are utilized by different viruses to fuel replication1. Depletion of the triglyceride-synthesizing enzymes diacylglycerol O-acyltransferase (DGAT) 1 and 2 reduced WNV, DENV, and ZIKV infection rates in knockdown cells, but only ZIKV titers were decreased. In contrast, depletion of the cholesterol-esterifying sterol O-acyltransferase (SOAT) 1 decreased WNV, TBEV, DENV and ZIKV infection rates and titers. Since we observed the strongest phenotype for ZIKV, we focused on studying the role of DGAT1/2 and SOAT1 in ZIKV infection.
In perspective of finding new druggable targets, we were curious if pharmacological inhibition of either enzyme could also reduce ZIKV infection. DGAT1 activity is important for ZIKV in placenta models2 and the effect of DGAT1 depletion was strong, so we had certain expectations. However, ZIKV happily replicated in DGAT1 inhibitor-treated Huh7 cells (and later on in other cell lines), leaving us dissatisfied. During her postdoc time in the US, Eva discovered that DGAT1 interacts with the nucleocapsid protein of the hepatitis C virus (HCV) and this interaction (and DGAT1 activity) is crucial for efficient particle production3. For ZIKV, activity is not the key, but maybe interaction is? We analyzed a potential interaction of DGAT1 and the ZIKV capsid protein and this time, we were not disappointed. Both proteins co-localized and interacted with each other, explaining why DGAT1 activity might be dispensable for ZIKV infection.
Regarding the role of SOAT1 activity, I have to be honest: If I only had looked at the viral titers, I would not have been convinced that enzyme activity is important at all. The results displayed a high variance and only a slight reduction for one of the two inhibitors we tested. But the dose-dependent reduction in virus protein level and reduced ZIKV cytopathic effects convinced us not to drop the idea of SOAT1 as druggable host factor just yet.
ZIKV is mainly neurotropic and liver cells are known to detoxify and metabolize rather fast. Two arguments, among many, to try different cell culture models. We repeated our experiments in neural cell lines, retrospectively the decision that made the whole project successful. In contrast to Huh7 cells, inhibition of SOAT1 strongly reduced ZIKV infection in microglia and astrocytoma cells, primary cells of ZIKV infection. At this point, I was ready to write the manuscript, but Eva suggested to additionally use a more sophisticated model: Our 3D cerebral organoids that my former colleague Vinicius was working on at that time. Needless to say, substantiating the effect of SOAT1 inhibition in this complex model was like the icing on the cake.
You add a substance to a cell and the virus stops multiplying. That´s great, but as a scientist the question is always: Why? What happens to the virus? Systematically, we analyzed the viral replication steps in terms of SOAT1 inhibition. While entry and RNA replication were still intact, protein level were lower, leading us to thinking we might be dealing with reduced production or spread. Indeed, we found less extracellular viral protein and RNA, and the specific infectivity of produced virions was lower. Thus, SOAT1 activity is important for ZIKV morphogenesis.
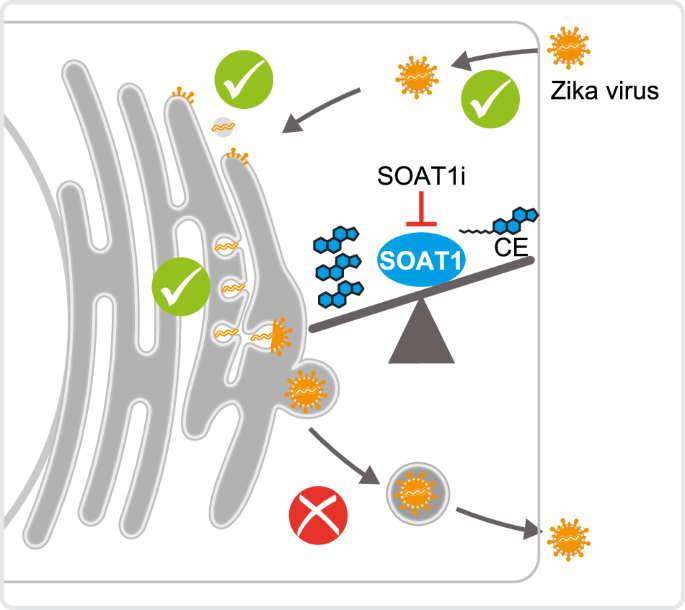
Now to the final question: How? What is blocking virus replication? Inhibiting SOAT1 blocks cholesterol ester synthesis. Are lower cholesterol ester levels detrimental for the virus? Or do we have an accumulation of the SOAT1 substrate cholesterol that affects virus replication? No to the first question, yes to the second. Supplementation with cholesterol ester did not rescue ZIKV infection in SOAT1 inhibitor-treated cells, but we observed a strong accumulation of free cellular cholesterol. Even though cholesterol is required for replication, do high levels tamper with ZIKV infection? To test this hypothesis, we added cholesterol during and post virus entry and observed a strong reduction in ZIKV infection, suggesting that too much cholesterol negatively affects ZIKV (Figure 1).

To sum up, we observed that blocking the activity of the cholesterol-esterifying enzyme SOAT1 reduces Zika virus infection. This is likely due to an imbalance between cholesterol and cholesterol ester that creates a disadvantageous lipid environment for virus particle morphogenesis.
References
- Herker E. Lipid Droplets in Virus Replication. FEBS Lett, (2024).
- Chen Q, et al. Metabolic reprogramming by Zika virus provokes inflammation in human placenta. Nat Commun 11, 2967 (2020).
- Herker E, et al. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat Med 16, 1295-1298 (2010).
Figures are modified from the original publication.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Your space to connect: The Myeloid cell function and dysfunction Hub
A new Communities’ space to connect, collaborate, and explore research on Clinical Medicine and Cell Biology!
Continue reading announcementRelated Collections
With Collections, you can get published faster and increase your visibility.
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026
DNA repair and human disease
Publishing Model: Hybrid
Deadline: Oct 31, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in