Key activity descriptors of nickel-iron oxygen evolution electrocatalysts in the presence of alkali metal cations
Published in Chemistry
Alkali metal cations (Li+, Na+, K+, Rb+, Cs+) are known to impact the activity of catalysts for various catalytic reactions. In this work, we investigate the influence of electrolyte cations on the activity of the nickel-iron oxyhydroxide catalyst, NiFe(OOH), one of the most promising catalysts for the oxygen evolution reaction (OER) in alkaline pH. Electrocatalytic OER (alias ´water-oxidation´) is the most energy demanding step in production of hydrogen from water. The transfer of four electrons and four protons to complete the catalytic cycle and to release O2 gives rise to what is generally recognized as three oxygenated “surface” intermediates (*OH, *O, *OOH), where tuning the activity involves changing the relative stability of these. Electrolyte cations have been found to modify the adsorption energies of the intermediates via non-covalent interactions. Small cations, with a higher Lewis acidity, are known to alter the stability of the intermediates in a detrimental manner. However, the OER activity does not always follow the properties predicted from the cation size, where unexplained deviations are often observed. In this study, we were driven to find an explanation for these discrepancies.
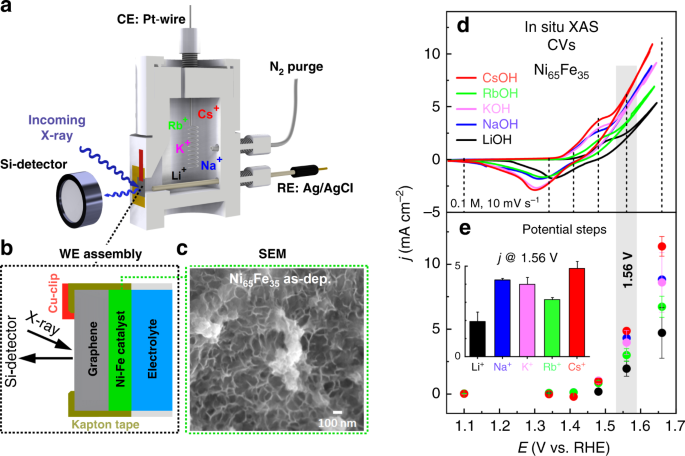
The source of cations in our study are alkali hydroxides (e.g. LiOH). Since the Lewis acidity decreases from small to large cations, the basicity of the corresponding hydroxide increases (predicted from their pKb values). Alkali hydroxides containing larger cations (i.e. CsOH) therefore dissociate to a larger extent and contribute to a higher OH- concentration, and thus a higher pH. We extended our investigations to a wider pH window to more easily catch this effect, since the pH differs more significantly between small and large cations at higher molar concentrations. As predicted, the OER activity indeed follows the variations in electrolyte pH, which also differs from the trend predicted from the cation size. When the activity was plotted against the pH instead of the nominal concentration, the otherwise large differences between the cations almost entirely vanished (see Figure 1). The pH takes into account not only the basicity of the alkali hydroxide but also other sources of error impacting the actual concentration of OH-, which contribute to unpredictable variations in the pH. Since the OER activity of the Ni-Fe(OOH) catalyst is sensitive to the electrolyte pH, the activity will respond thereafter. It should be noted that we faced great challenges with determining the pH in LiOH, and we tested several alternative approaches. The accuracy is therefore lower in LiOH, which could be the reason why the activity somewhat deviates from the linear trend.
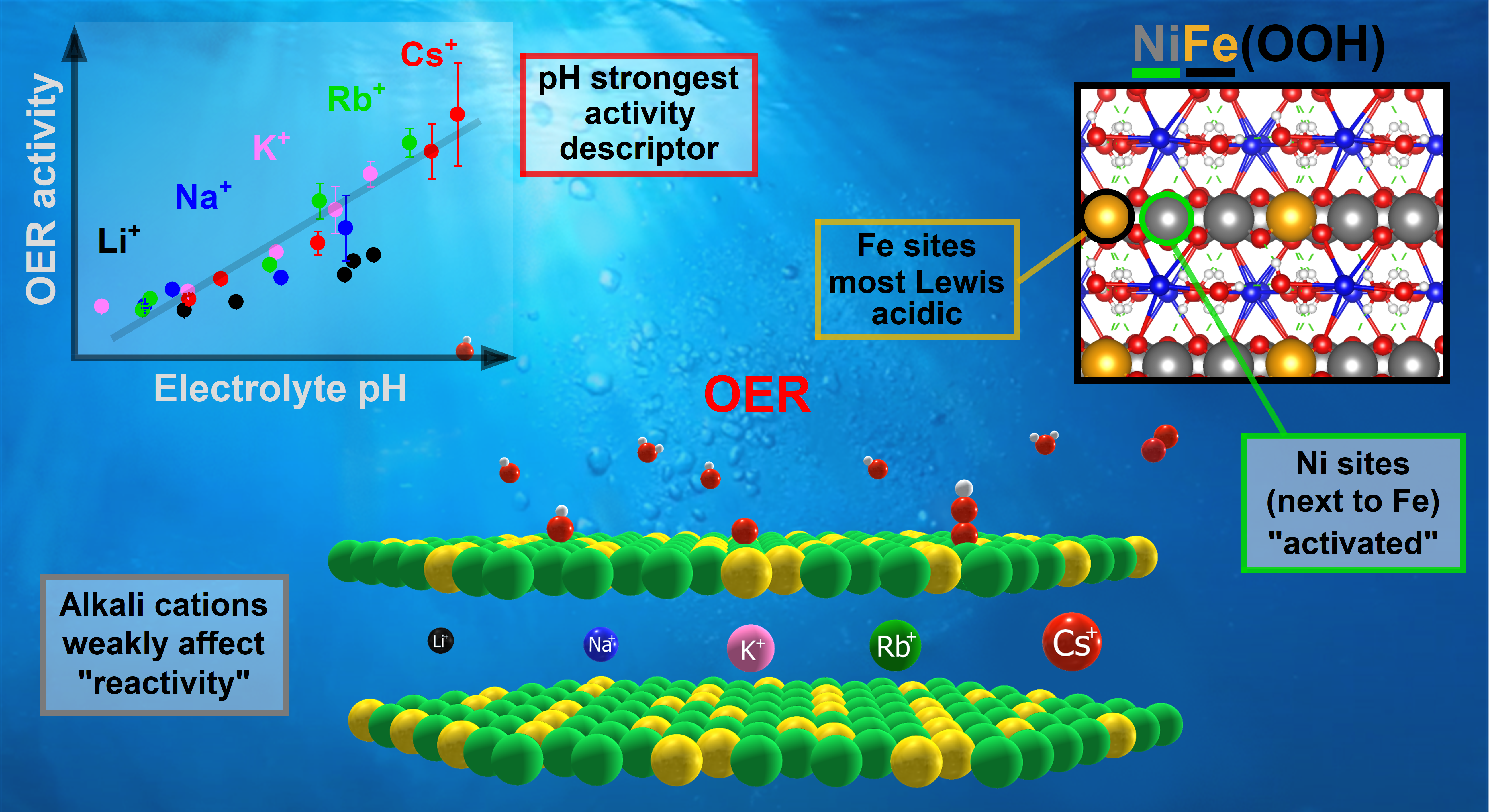
Figure 1. The impact of alkali metal cations on the OER activity. The influence of alkali metal cations (LiOH, NaOH, KOH, RbOH, CsOH) on the OER activity of the Ni-Fe(OOH) catalyst. The pH generally increases from small to large cations due to an increase in the basicity of the corresponding alkali hydroxide. However, other sources of error contributes to deviations in the nominal OH- concentration, whereby the electrolyte pH not always strictly follows the trend in cation size. Since the OER activity of the Ni-Fe catalyst is sensitive to the pH, the OER activity follows the variations in pH, which cannot simply be predicted from the size of the cation. DFT-derived reactivity descriptors also support that cations have a negligible effect on the Lewis activity and basicity (and thus the "reactivity") of the lattice sites of the Ni-Fe(OOH) catalyst, which supports an indirect pH effect. We also confirm that Fe-dopants do affect the reactivity descriptors more strongly, which is in line with both previous experimental and theoretical findings.
To further support our observations, we investigated a series of theoretical DFT-derived reactivity descriptors that correlate with the Lewis acidity and basicity of the catalyst lattice sites (in this case "edge" sites); Ni, Fe, and O. Our results show that alkali metal cations do not greatly affect these reactivity descriptors in contrast to Fe-dopants (in NiOOH) which impact all the reactivity parameters more significantly. Although small, there might be a "specific” cation effect to some extent, however, we judge that this effect is unlikely to account for the entire effect on the catalytic activity, which according to our experimental data is dominated by the electrolyte pH. Our study therefore brings attention to an important activity descriptor - the electrolyte pH - in studies involving alkali metal cations in alkaline OER.
To read more about our work: Görlin, M. et al. Key activity descriptors of nickel-iron oxygen evolution electrocatalysts in the presence of alkali metal cations. Nat Commun 11, 6181 (2020). https://doi.org/10.1038/s41467-020-19729-2
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
Nice attempt. Congratulations!!