

The development of equipment for analyzing the base sequence of DNA has been remarkable. Today, it is possible to obtain the entire genome sequence of a single human being from a single device. However, the resulting sequences are a collection of short fragments (short reads) of one or several hundred bases, like pieces of a jigsaw puzzle. Reordering these pieces to reconstruct the chromosomes is possible but requires enormous calculations.
If you only want to check for the presence or absence of mutations from the sequence information, you can detect them by searching for parts of the base sequence in short reads that differ from the whole genome sequence of the target without reordering the pieces. However, since the obtained sequence also contains errors, it is necessary to determine whether the mutation actually occurred or if it is due to an analysis error of the instrument, based on the frequency of detection events.
In the case of a substitution mutation where one base in the sequence is replaced, it is possible to detect the mutation site in the whole genome sequence simply by removing the noise. It is also possible to detect insertions and deletions that are sufficiently smaller than the length of the analyzed fragment, for example, a few bases. However, insertions and deletions longer than the length of the fragment are difficult to detect directly.
For example, if a sequence with a deletion from 31,423,139 to 31,427,498 of the genome position is analyzed by BLAST, the result is output in two parts. If they are output side by side, you will notice the deletion, but if they appear in a large number of results, it is difficult to determine that they are deletions (Figure 1).

I struggled with the sequence every day. One day, the idea occurred to me: what would happen if I aligned the reference sequence against the short read sequence instead of the short read sequence against the genome reference sequence?
However, the reference sequence of the genome is huge. In addition, reads are short but enormous in number. There was no program anywhere to align genome reference sequences for short reads.
Therefore, I decided to create a program from scratch to align a huge size of genome sequences for a huge number of short reads. I overcame the problem of exceeding memory and disk capacity with various ingenuities and finally succeeded in developing a practical program.

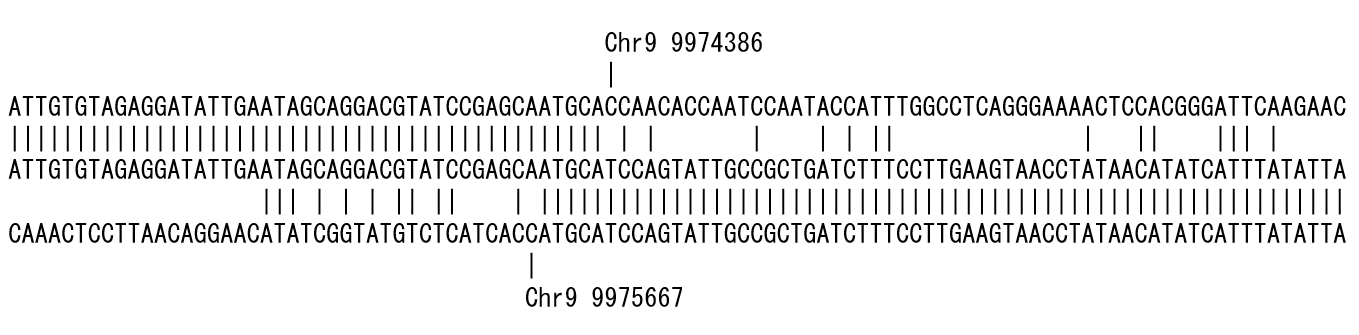
Figure 2 is the result of placing the short read sequence in the middle row and aligning the genome reference sequence from the 5' end side in the upper row and the 3' end in the lower row. The position where the mismatch occurred is shown up and down. In this example, a deletion of approximately 1.3 kb could be detected.
I named this alignment style ‘bidirectional alignment’. Since the alignment proceeds from both ends of the short read to detect the mismatch, i.e., polymorphism, the position where the polymorphism is detected, the program is called polymorphic edge detection (PED).
Today, devices have been developed that allow you to read longer fragments. Although it is easier to detect insertions and deletions by using long fragment data, I believe that it is an effective way to utilize the enormous amount of short-read data that has been accumulated so far. The bidirectional alignment will reveal new aspects from the short reads.
Miyao, A., Kiyomiya, J.S., Iida, K. et al. Polymorphic edge detection (PED): two efficient methods of polymorphism detection from next-generation sequencing data. BMC Bioinformatics 20, 362 (2019). https://doi.org/10.1186/s12859-019-2955-6
Follow the Topic
-
BMC Bioinformatics
This is an open access, peer-reviewed journal that considers articles describing novel computational algorithms and software, models and tools, including statistical methods, machine learning and artificial intelligence, as well as systems biology.

Related Collections
With Collections, you can get published faster and increase your visibility.
Computational methods in paleobiology
BMC Bioinformatics is welcoming submissions to our Collection on Computational methods in paleobiology.
The interdisciplinary field of paleobiology enables researchers to reconstruct evolutionary histories, model population dynamics, and explore the multifactorial influences that shaped life in the past. Paleobiology includes, but is not limited to, macrofossils, microfossils, and ancient proteins, DNA and RNA from both fossils and environmental sources. For instance, with the rapid growth of paleogenomic datasets, novel computational approaches are essential for extracting insights from this fragmented ancient data.
This Collection welcomes submissions on the development of new computational and/or statistical approaches for paleobiology. We encourage contributions that highlight innovative methods for analyzing ancient DNA, modeling evolutionary processes, integrating heterogeneous datasets, and visualizing temporal and spatial patterns.
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer-review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Jan 23, 2027
Cell tracking
BMC Bioinformatics is welcoming submissions to our Collection on Cell Tracking.
Cell tracking is a technique used to monitor and analyze the movement and behavior of cells over time, allowing the study of cellular behaviors, dynamics, and interactions within various biological contexts. Advanced bioinformatics tools play a vital role in analyzing cell tracking data. They help identify cell movement patterns and understand their biological implications. These tools are particularly relevant when processing large datasets and when investigating cell cycles.
This Collection welcomes submissions on the development of new computational and/or statistical approaches for cell tracking. We encourage contributions detailing methods for detecting and characterizing cell movements to better understand cell migration and behavior.
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer-review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Sep 23, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in