Mapping the functional proteome landscape of Escherichia coli
Published in Microbiology
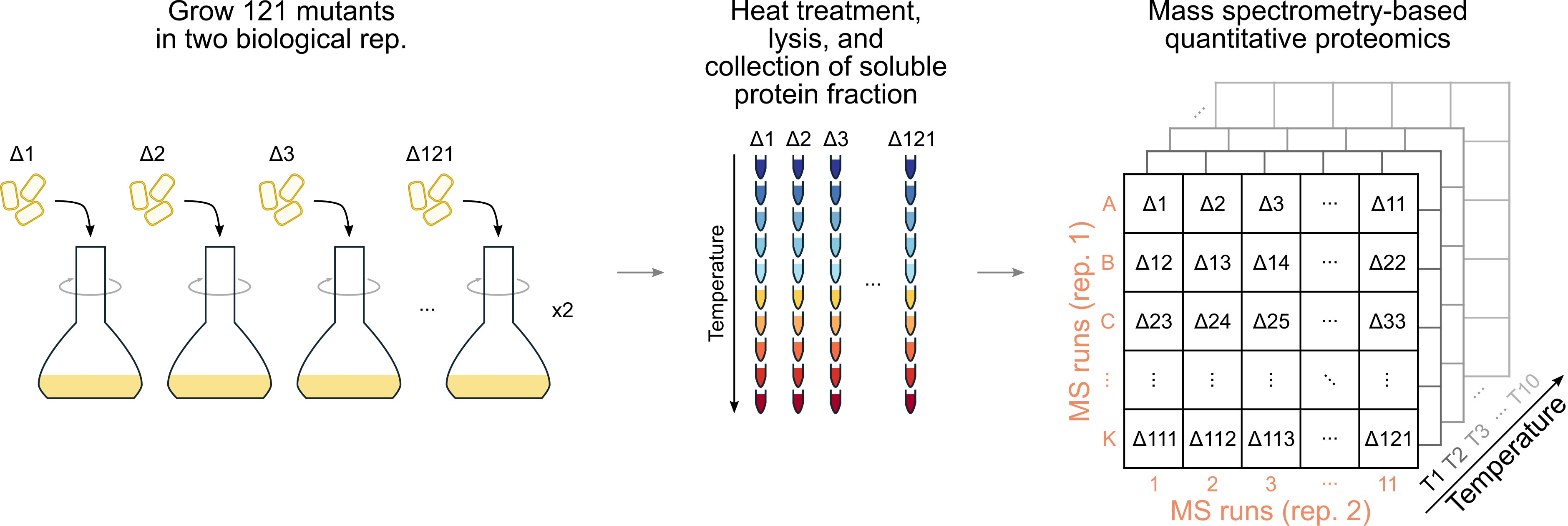
One of the main goals of molecular biology is to identify the function, regulation, and interactions of genes and their products. Our group has developed a proteomics approach, termed thermal proteome profiling (TPP), that relies on the principle that protein thermal stability is affected by protein interactions, such as those with drugs, metabolites, nucleic acids, or other proteins. Since it can be performed in living cells, TPP offers an unprecedented view of the state of the cell. We have recently adapted the protocol to work in bacteria and immediately got curious on how scalable and powerful this technique could be at characterizing the proteome of an organism. For this, we were inspired by high-throughput reverse genetics approaches, and decided to profile proteome-wide abundance and thermal stability for more than one hundred knock-outs of genes targeting a variety of cellular processes—the idea being that we would perturb the proteome in multiple ways.
This work is now published in Nature and all the data can be easily explored in multiple ways at https://ecolitpp.shiny.embl.de/.
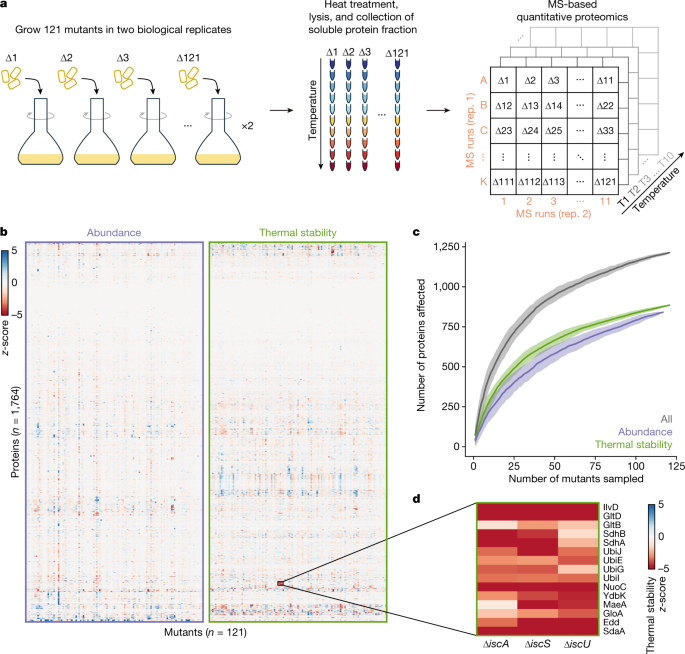
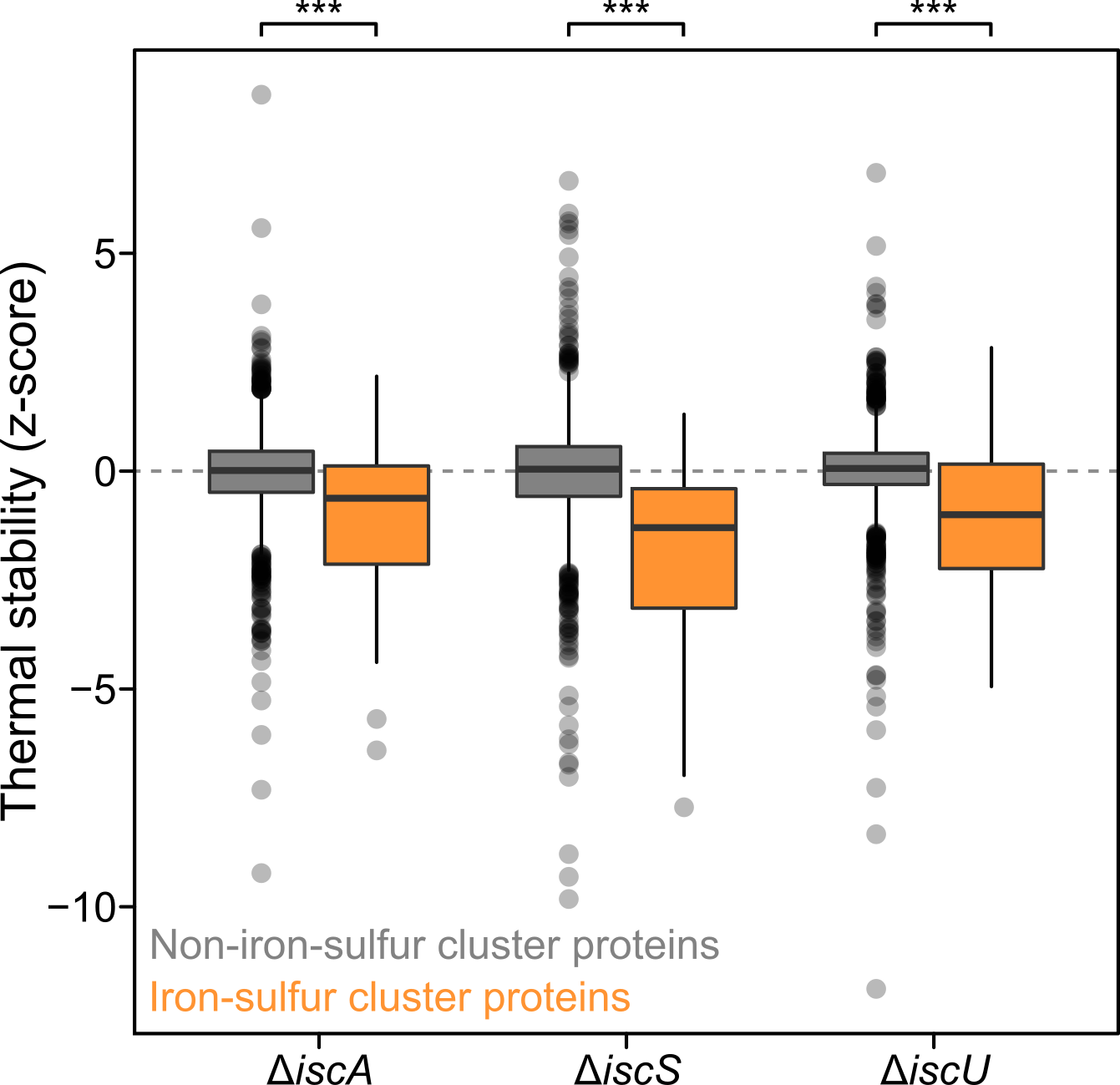
One way to look at the data is to look directly at the effects of a gene deletion on the proteome to infer its function. For example, in mutants of iron-sulfur cluster biosynthesis (ΔiscA, ΔiscS, ΔiscU) many proteins that bind this co-factor where thermally destabilized, since the cells cannot produce it. We also observed that enzyme thermal stability was correlated with the levels of the metabolites that bind it, allowing us to monitor the metabolic state of the cell.
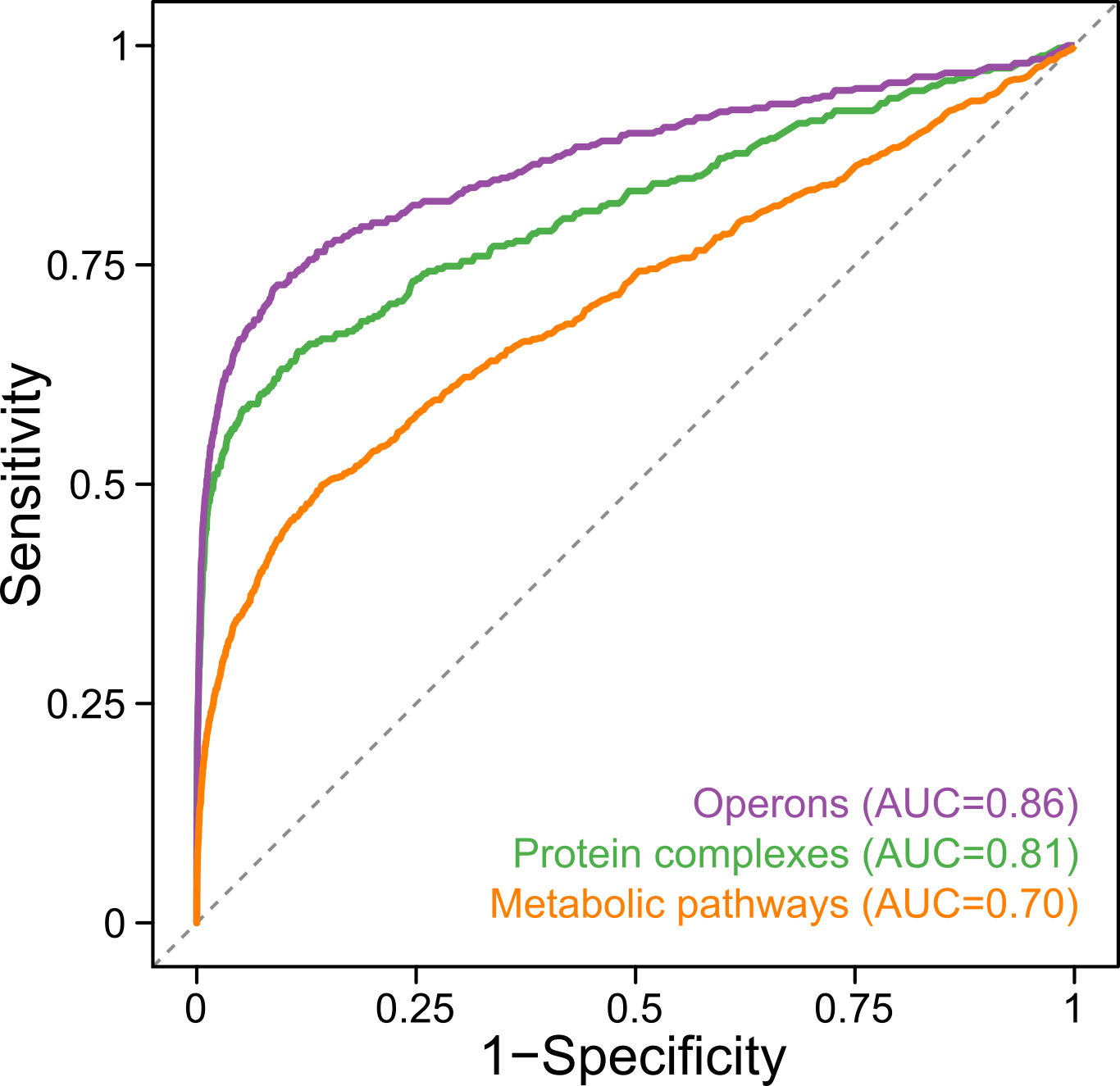
Another way to look at the data is to ask which proteins behave similarly in all the mutants studied, because proteins that have related functions should have similar interactions in the different mutants. This is a powerful approach, since it allows us to look beyond the one hundred gene knock-outs that we profiled, and into the 1,800 proteins that we can detect. When we compared this analysis to previously annotated functionally-related proteins, we saw that we were quite good at recapitulating what is already known, but we also suggest possible new links for the function of hundreds of proteins, including many of completely unknown function.
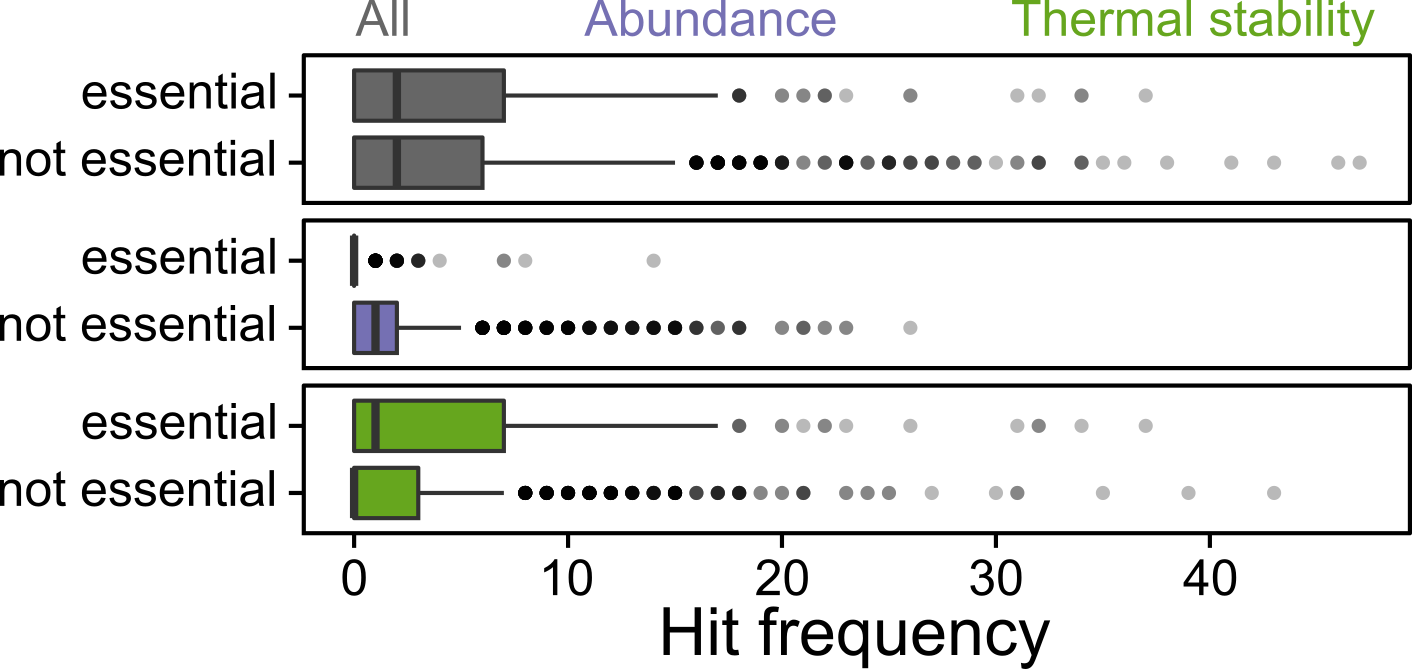
An interesting finding from our study was that proteins that are essential for life (i.e., cannot be deleted and thus are difficult to study with genetics approaches) rarely changed in their abundance, but commonly changed in their thermal stability. We showed that some of these proteins were more important in mutants that had changes in their thermal stability, since those mutants could not tolerate their downregulation.
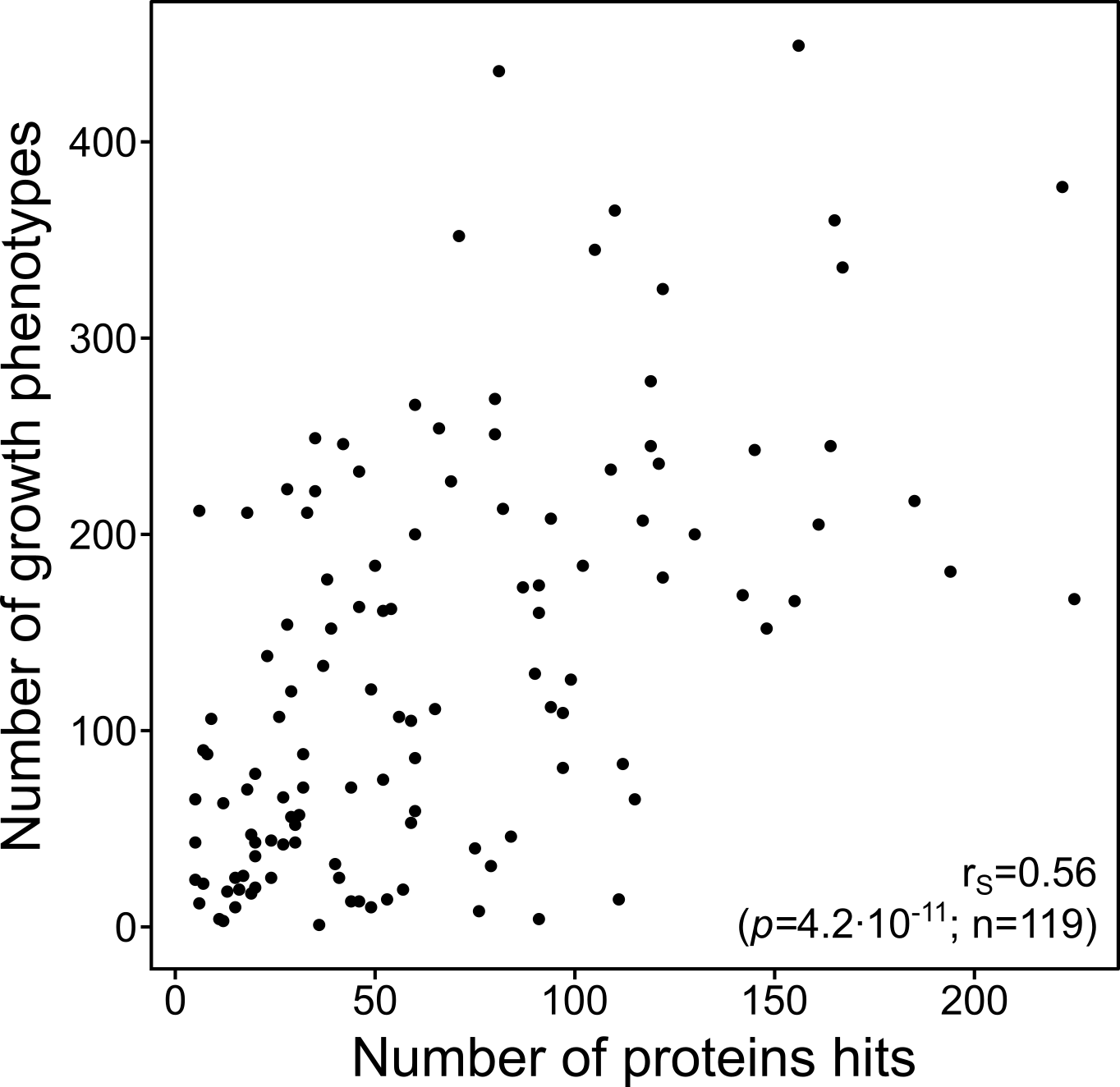
Finally, we compared our data with previously acquired growth phenotypes for these mutants. In general, we saw that mutants that had a larger proportion of the proteome affected were more sensitive to any external perturbation . In addition, we could explain some phenotypes that were caused by changes on the proteome that go beyond the deleted gene. For example, we saw that low levels of the multidrug transporter MdtK were responsible for the sensitivity of some mutants to the antidiabetic drug metformin—these mutants target processes that are not directly related with the regulation of MdtK.
It will now be exciting to expand this approach to other microbes. With over 20,000 bacterial species identified by now, and an increasing number of them being culturable and genetically-tractable, many proteins of unknown function lie in waiting characterization.
Read the full paper at: https://www.nature.com/articles/s41586-020-3002-5
Follow the Topic
-
Nature
A weekly international journal publishing the finest peer-reviewed research in all fields of science and technology on the basis of its originality, importance, interdisciplinary interest, timeliness, accessibility, elegance and surprising conclusions.

Related Collections
With Collections, you can get published faster and increase your visibility.
Carbon Dioxide Removal
Publishing Model: Hybrid
Deadline: Jan 16, 2027

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in