Molecular dynamics from first principles
Published in Chemistry

Background - Force Fields, molecular simulation, mixtures and ensembles
The physicist Richard Feynman said, “all things are made of atoms, and that everything that living things do can be understood in terms of the jigglings and wigglings of atoms” 2. Once the total energy (the Hamiltonian) of a system is properly determined, physics enables one to derive the system’s properties. Since, in principle, quantum mechanics (QM) tells us how to obtain this energy for a system of atoms and molecules, one may be tempted to conclude that modelling ‘every living thing’ is a solved task. In practice, however, no one knows how to use quantum mechanics to accurately model something as commonplace as a lump of sugar dissolving in a cup of tea. The fastest ab-initio methods (density functional theory, DFT) scale as the number of atoms N to the power of ~5, and the most faithful ones as ~N^7. Modelling your morning tea requires a system of at least 128 atoms and, as a multitude of states must be visited to determine the entropic contribution, requiring at least 10^5 energy evaluations. Simulations of biological molecules, which can contain hundreds of thousands of atoms and much longer time-scales 3 than solvation, appear completely intractable, no matter how much faster our computers will be.
To overcome this intractable computational limit, scientists have replaced quantum energies and forces with much faster and immensely better-scaling (N log(N)) scalar approximations of QM called Force Fields (FF), which are propagated via Newton’s laws of motion (Molecular Mechanics or MM). One may simulate the whole ensemble of molecules in MM or, when better accuracy is required at some region such as a catalytic site, stitch MM for most of the atoms with QM for a small accuracy-demanding subset. Recognition of the increasing importance of these multiscale simulations led to the award of the 2013 Nobel Prize in Chemistry to Martin Karplus, Michael Levitt (one of the current authors), and Arieh Warshel “for the development of multiscale models for complex chemical systems” 4.
The original Force Fields derived many of their model parameters by fitting to available experimental data. For several reasons 9 it is far more advantageous to define the approximation of the true QM energies by FF by actually fitting the FF to QM: one most obvious reason is the ability to model molecules which have not yet been synthesized. However, the pioneers of molecular modeling did not fully rely on QM data partially because of the prohibitive cost of quantum computations in the 1960-70’s. In the 50 years since the original work, the ability to obtain high quality QM data to set the FF parameters has increased by over 9 (!) orders of magnitude. Yet, to this day, the current work-horses of biological molecular simulations are parameterized partially by fitting to experimental data such as densities and heats of vaporization. One of the reasons for this anachronism is that the substitution of Quantum Mechanics by Force Fields introduces significant complexity into the approximation: An atom that previously only needed its place in the periodic table specified, now must be classified further: a carbon becomes an aliphatic, aromatic or perhaps a carboxylic one. Moreover, each ‘type’ acquires a list of properties: e.g. partial charges, Van der Waals radii, dipole moments, polarization constants, and so on. In an essay ‘On Exactitude in Science’ Jorge Louis Borges derides a map of a 1:1 scale, saying dryly: "[S]ucceeding Generations... came to judge a map of such Magnitude cumbersome…” 5. A similar opinion is offered by Lewis Carrol: "the farmers objected: they said it would cover the whole country, and shut out the sunlight !” 6. It is therefore critical to find a level of model complexity that is both tractable but also permits a sufficiently high agreement with the quantum calculations.
 Fig. 1: The final Gibbs free energy (blue) is really a sum and difference of many components (green and yellow).
Fig. 1: The final Gibbs free energy (blue) is really a sum and difference of many components (green and yellow).
The level of complexity depends on the desired accuracy of prediction of molecular simulations. This is often stated to be 0.5 kcal/mol, which is roughly the level of thermal noise at room temperature and pressure. For example, a decrease in protein-ligand Gibbs free energy of binding by 0.5 kcal/mol will result in ~2X increase in ligand binding, which is an improvement a medicinal chemist will find worth pursuing. To predict the final number reliably, however, the actual internal accuracy of models has to be much higher because the total energy change arises from the addition and cancellation of many large components. These components come from the interaction between molecules, as well as from the entropic contributions which are of similar magnitude to the enthalpic ones. An intuitive illustration of getting the final answer as a sum of opposing larger components for three states of matter is illustrated in Fig. 1.
Our research and results
In Pereyaslavets et al 2022 1 we report on the results achieved by a Force Field parameterized entirely by QM calculations and without any reliance on experimental results. The Arrow FF is capable of describing the liquid state to within chemical accuracy. The level of complexity required is quite high: 1) the FF is polarizable, allowing transferability from gas to liquid phase, 2) the FF models charge penetration effects well, 3) the FF describes the shapes of atoms anisotropically, and 4) ring polymer MD is used to model the Nuclear Quantum Effects (NQE) 7.
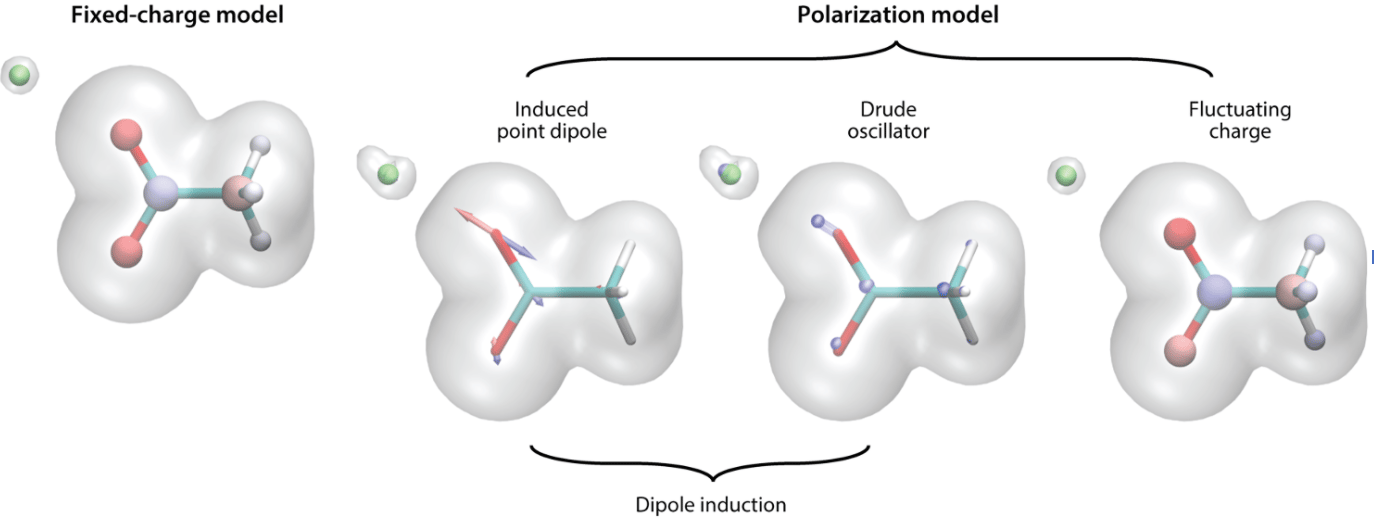 Fig. 2: Electron cloud: Fixed charge versus polarization models (From Jing et al 2019, reproduced with permission) 8
Fig. 2: Electron cloud: Fixed charge versus polarization models (From Jing et al 2019, reproduced with permission) 8
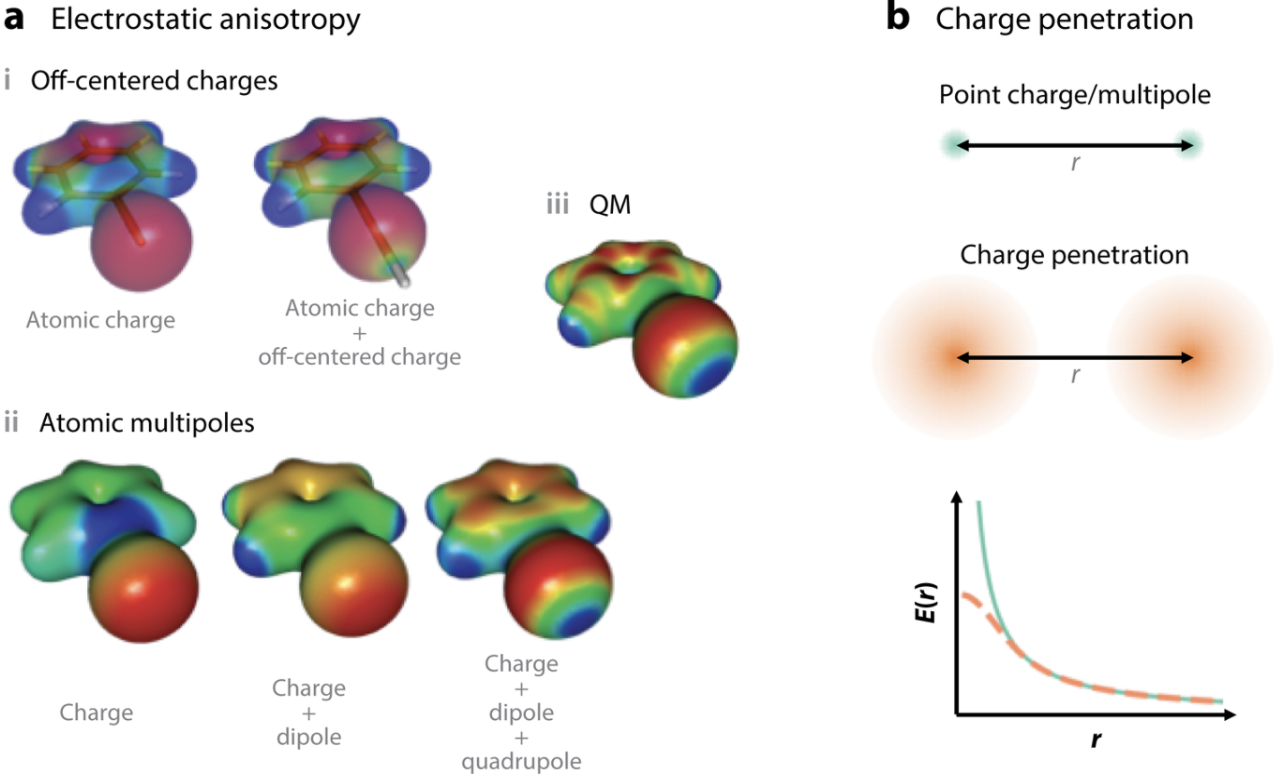
Fig. 3: Electrostatic anisotropy and charge penetration. The Arrow FF also uses anisotropy in its exchange-repulsion and induction components. (From Jing et al 2019, reproduced with permission) 8
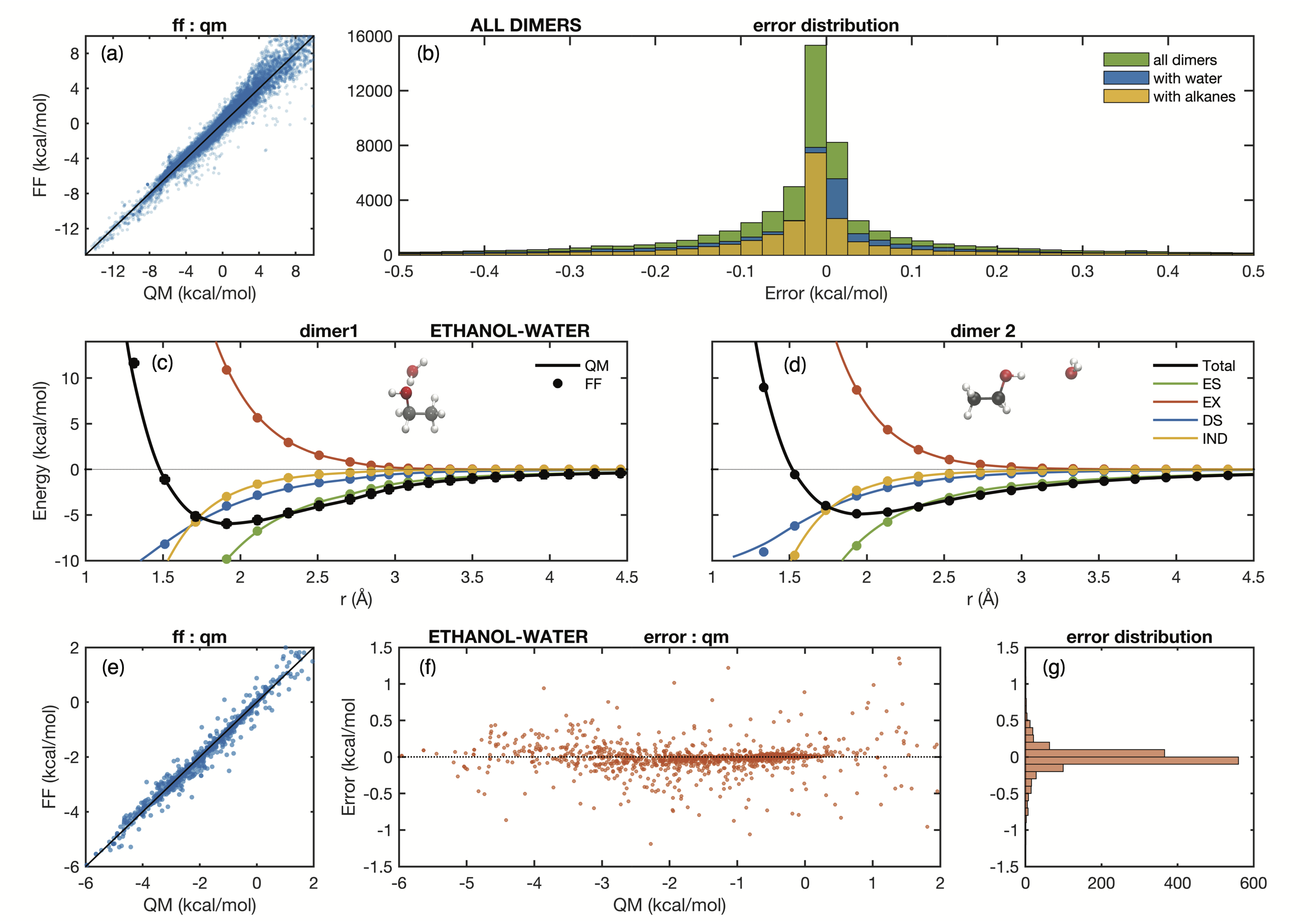
As a result of the included complexity Arrow FF agrees very well with the QM energies. A plot of this agreement for dimer (2-body) energies is shown in Figure 4:
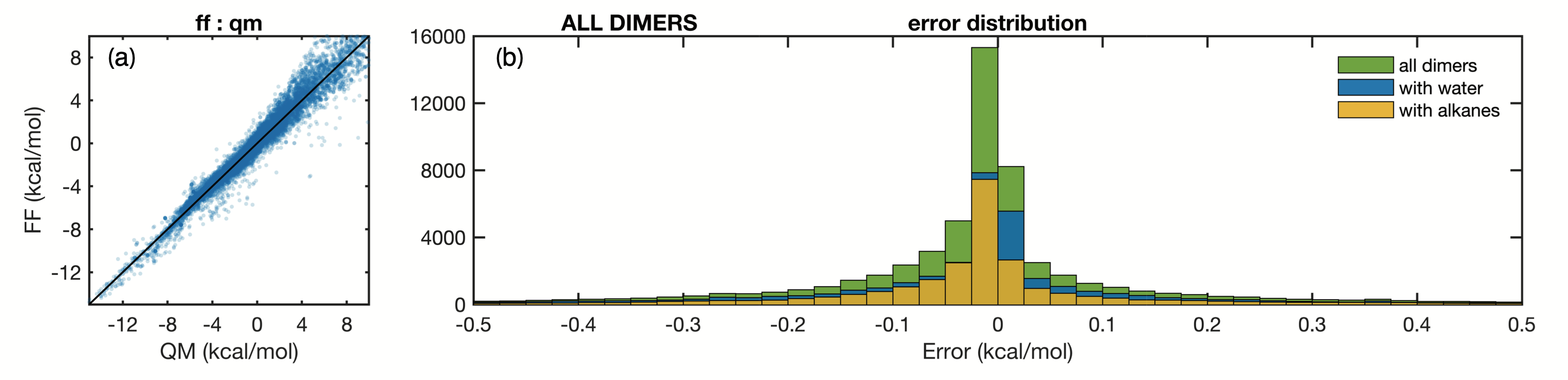
Fig. 4: The agreement between QM and FF dimer energies (2-body) in Arrow FF is very good. Similar fits are achieved for many-body contributions to the total energy.
At the same time, the level of complexity is manageable. As a result, the model is able to cover all major known neutral functional groups and solvents, and retain the ability to compute many nanoseconds per day. We confirm our previous finding 9 that treating the motion of the nuclei in a quantum manner is necessary for obtaining the desired accuracy of free energy predictions:
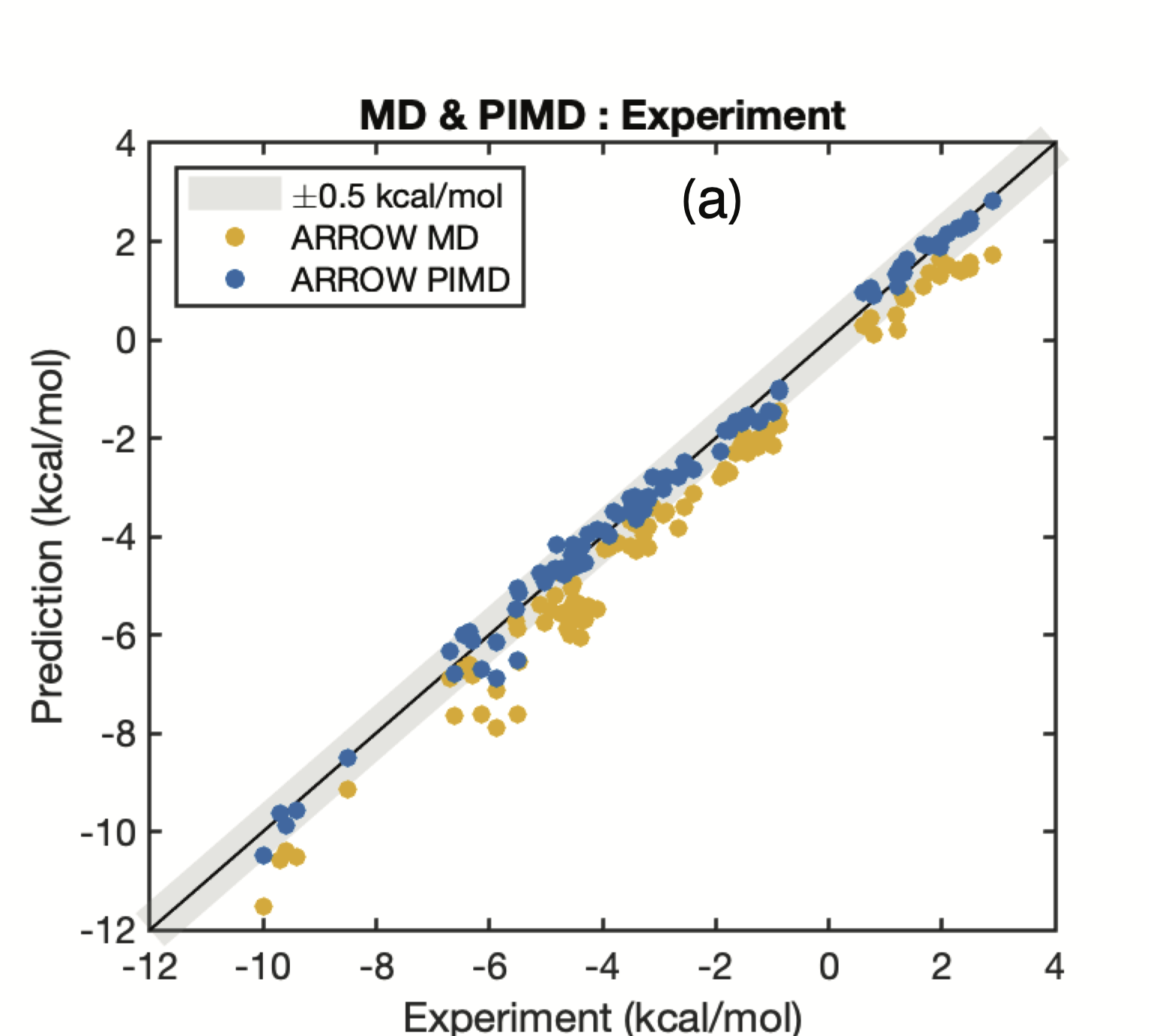
Fig. 5: The inclusion of Nuclear Quantum Effects (NQE) brings the predictions of all the mixtures into excellent agreement with experimental values 1
The prediction results are satisfyingly accurate. Most of the functional groups’ computed free energies of hydration and solvation in Cyclohexane (CHEX) are well within chemical accuracy:
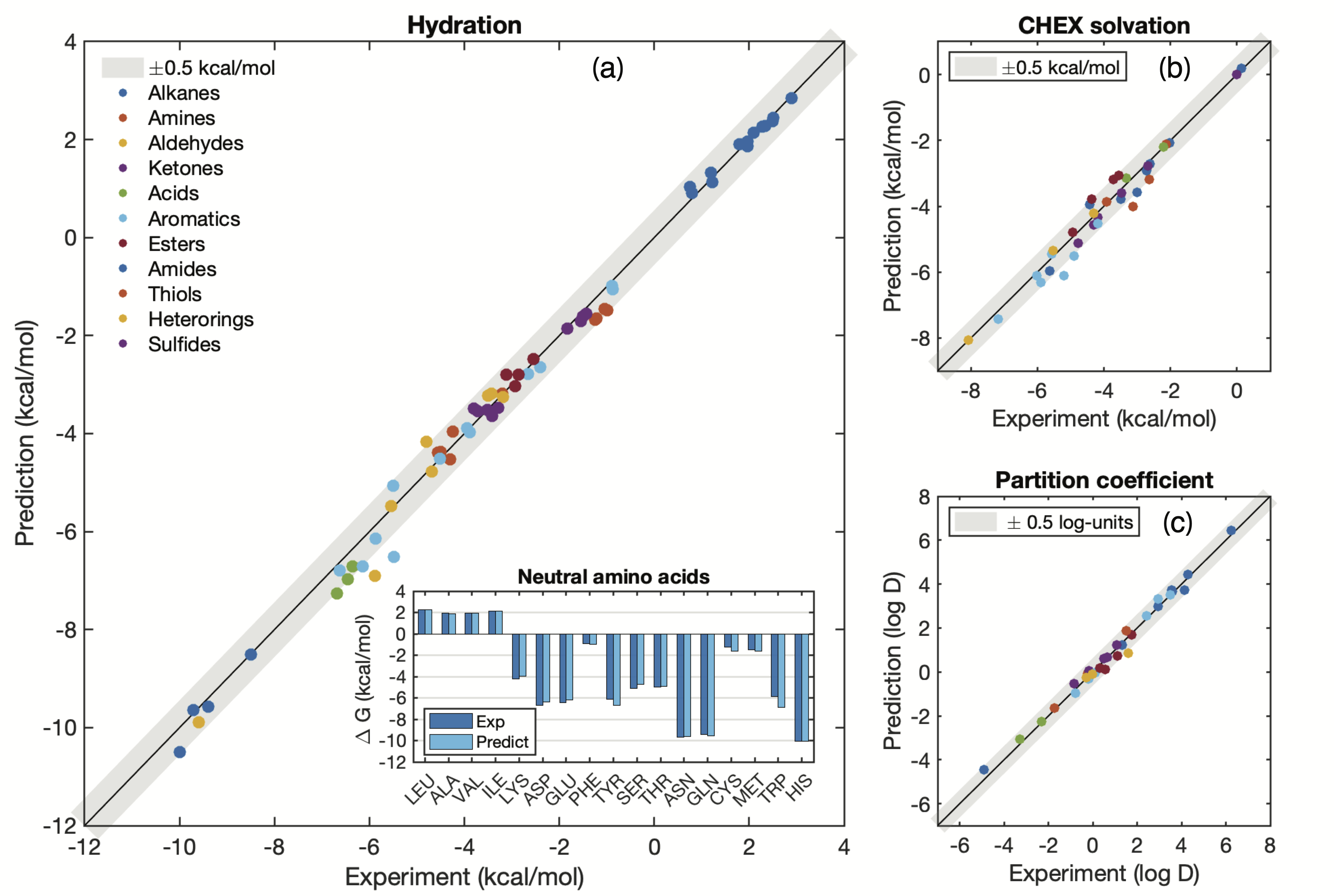
Fig. 6: Predicted versus experimental free energy of hydration (a), solvation in cyclohexane (b), and water/cyclhexane partition coefficients, for a diverse set of compounds. The grey bar is + 0.5 kCal/mol 1.
We achieved the long-standing goal of using an ab-initio parameterized model to accurately predict the behavior of a wide range of neutral molecules in the liquid phase. Whether this kind of model will be sufficient for describing the more challenging protein-ligand systems and ionic liquids remains to be seen and is the subject of research both by our group and by many others. Please visit research.interxinc.com for collaboration requests and further information.
References
- Pereyaslavets L, Kamath G, Butin O, et al. Accurate determination of solvation free energies of neutral organic compounds from first principles. Nature Communications. 13, 414, 2022..
- Feynman RP, Leighton RB, Sands ML. The Feynman lectures on physics. New York, NY: Basic Books; 2010.
- Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature (London). 2007;450(7172):964-972. https://www.ncbi.nlm.nih.gov/pubmed/18075575. doi: 10.1038/nature06522.
- The Nobel Prize in Chemistry 2013. https://www.nobelprize.org/prizes/chemistry/2013/summary/ Web site. . Accessed 01/17/, 2022.
- Borges JL, Hurley A. Collected fictions. New York: Penguin Books; 1999. http://www.gbv.de/dms/bowker/toc/9780670849703.pdf.
- Carroll L. Sylvie and Bruno. London, New York: Macmillan; 1889.
- Craig IR, Manolopoulos DE. Quantum statistics and classical mechanics: Real time correlation functions from ring polymer molecular dynamics. The Journal of chemical physics. 2004;121(8):3368-3373. https://www.ncbi.nlm.nih.gov/pubmed/15303899. doi: 10.1063/1.1777575.
- Jing Z, Liu C, Cheng SY, et al. Polarizable force fields for biomolecular simulations: Recent advances and applications. . . doi: 10.1146/annurev-biophys-070317-.
- Pereyaslavets L, Kurnikov I, Kamath G, et al. On the importance of accounting for nuclear quantum effects in ab initio calibrated force fields in biological simulations. Proceedings of the National Academy of Sciences - PNAS. 2018;115(36):8878-8882. https://www.jstor.org/stable/26531200. doi: 10.1073/pnas.1806064115.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in