Molecular mechanism of specific HLA-A mRNA recognition by the RNA-binding-protein hMEX3B to promote tumor immune escape
Published in Cell & Molecular Biology and Immunology
Lots of progress has been achieved in the field of cancer immunotherapy. Immune checkpoints (ICs), including programmed cell death protein-1 (PD-1), programmed cell death ligand-1 (PDL1), and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), are a class of immune-modulating proteins involved in the negative regulation of the immune system. Immune checkpoint blockade (ICB) specific to immune checkpoint molecules has emerged as one of the most promising approaches in cancer immunotherapy. However, these durable responses are only limited to a subset of patients, and the mechanism behind why some people cannot respond to immunotherapy efficiently is still unknown. Understanding the molecular mechanisms of resistance to immunotherapy is critical for finding new strategies to overcome resistance to immunotherapy. T cells are crucial in the adaptive immune system and distinguish between healthy and tumor cells. Upon recognition of tumor-specific antigen fragments (peptides), activated T cells will contribute to the immune response. The major histocompatibility complex (MHC) molecules, or human leukocyte antigens (HLA) in humans, bind these antigen peptides to present them to T cells and be recognized by the T cell receptors (TCR) on the surface of T cell. This recognition event is the first step that leads to T cell activation preceding the immune response.
In 2018, Huang and colleagues used a kinome library screen to identify hMEX3B, an RNA-binding protein, as an essential regulator of melanoma resistance to PD-1 blockade immunotherapy. Low expression of hMEX3B in melanoma cells was strongly associated with response in a cohort of patients with melanoma treated with anti-PD1 checkpoint blockade. hMEX3B overexpression is associated with resistance to PD-1 blockade. This effect of hMEX3B was dependent on the endogenous expression of HLA-A and could be reversed by overexpression of exogenous HLA-A. The authors demonstrate that the hMEX3B disrupts HLA-A by binding the 3′UTR of its mRNA. This important work illustrated that antigen presentation is identified as a critical pathway in resistance mechanisms to immune checkpoint blockade and hMEX3B plays an essential role in this pathway. Even so, there still existed some unclear questions, such as how the hMEX3B binds to 3′UTR of HLA-A mRNA and how it regulates the expression of HLA-A. To answer these questions, we have done a structural biology study and function study for hMEX3B and HLA-A mRNA.
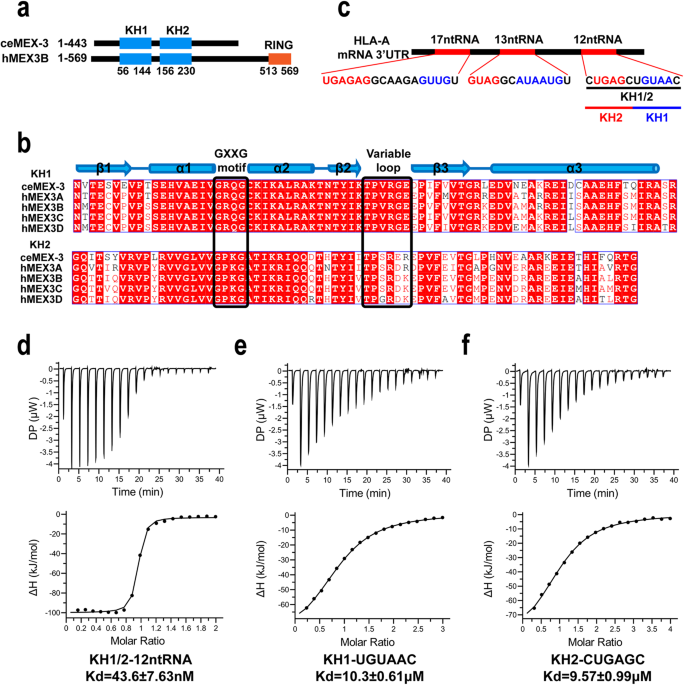
MEX-3 family proteins are RNA-binding proteins (RBPs). In mammals, four mex3 family genes encode four homologous proteins (hMEX3A ~ hMEX3D). All four hMEX-3 proteins comprise N-terminal two tandem KH domains, and the C-terminus, a typical C3HC4-type zinc finger RING domain, has E3 ubiquitin ligase activity. The KH domain is an RNA binding domain that exists in both eukaryotes and prokaryotes and contains three α-helices, three β-sheets and a conserved GXXG motif. Two KH domains recognize single-strand RNA substrates, a previous research from our laboratory reported the structures of individual KH domains of hMEX3C in complex with RNA fragment, which are the only reported hMEX-3 family protein-RNA complex structure. Based on the structural studies, the authors suggested the RNA substrate motifs recognized by hMEX3C and the bound pattern between the individual KH domains and the corresponding RNA substrates. In the current research, we combined the NMR and SAXS methods and demonstrated tandem KH domains of the hMEX3B in complex with the 3 ′UTR RNA fragment derived from HLA-A mRNA. We observed conformational change between the RNA-free and RNA-bound proteins. Furthermore, we determined two pairs of threonines and arginines (TRTR) on the KH domains’ variable loops, crucial in recognizing hMEX3B and RNA substrate. We investigated how TRTR mutation affects hMEX3B function and the downstream pathway both in vitro and in vivo.
Unlike its homolog in C. elegans, besides the N-terminal tandem KH domains that recognize HLA-A mRNA, hMEX3B also has a RING domain at its C-terminus, which has E3 ligase activity. The hMEX3B RING domain recruits the CCR4-NOT complex, through the interaction between RING and CNOT2/3 heterodimer, to the target mRNA recognized by the KH domains. After deadenylation, the target mRNAs are degraded by 5′-3′ exoribonuclease 1 and 2 through the exonuclease pathway. hMEX3B, as a negative effector of anti-PD-1 antibody therapy, reduces the HLA-A amount by degrading its mRNA to increase the immune escape ratio of the tumor cells from the T cell.
In summary, we detailedly and systematically examined the characteristics of how hMEX3B interacted with its substrate RNA, especially the necessity of two KH domains working spontaneously and coordinately. In particular, we narrowed the key residues that disrupted the interactions to TRTR, which may provide a new window leading to a potential gene therapy targeting hMEX3B, especially for patients who failed PD-1 antibody therapy.
I have a cross-disciplinary educational background in both computer science and biology. In the past five years, my main work has been to use interdisciplinary technical means to study the mechanism of mitochondrial dysregulation in degenerative diseases at a cross-scale and the study of cell in situ spatial structure biology at near-atomic resolution. My team has published 14 research papers in top journals such as Cell Discovery, Nature Cell Biology, Nature Communications, Communications Biology, and Advanced Materials in the past five years. My interest has mainly focused on: 1) combining computer modeling, elucidating the important role of multiple post-translational modifications of mitochondrial ribosomes in mitochondrial function and cell homeostasis maintenance, as well as the molecular mechanisms of information communication and bidirectional regulation between mitochondria and the two organelles of the nucleus; 2) A novel fluorescent carbon dot specifically targeting RNA granules was synthesized from scratch and applied to electron microscopy imaging and fluorescence imaging, elucidating the material exchange process among multiple membraneless organelles within cells; 3) We comprehensively utilized synthetic chemistry, cell biology, and structural biology to systematically analyze the molecular mechanisms of tumor cell escape from T cell killing and negative regulation of PD-l immunotherapy.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026
DNA repair and human disease
Publishing Model: Hybrid
Deadline: Oct 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in