Mutations may offer clues to cancer development in systemic sclerosis
Published in Protocols & Methods, Cell & Molecular Biology, and Genetics & Genomics
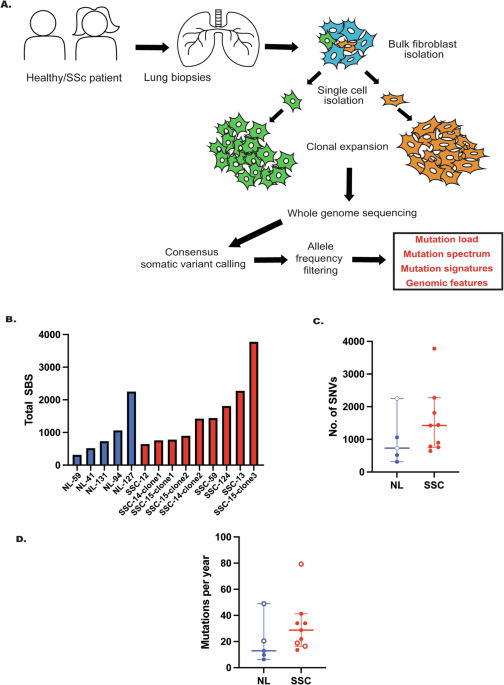
Systemic sclerosis (SSc), or scleroderma, is an idiopathic, autoimmune disease that disproportionately affects women, largely in late adulthood. The disease in typically characterized by excessive extracellular deposition of collagen, a central component of connective tissues such as muscles, ligaments, and skin and bones. This phenomenon often leads to hardening of these tissues, which lead to patients presenting with numerous complications such as tingling/numbness in extremities, hypertension, and respiratory problems. Despite recent therapeutic advances, there is so far no cure for systemic sclerosis.
Remarkably, up to one in five patients develop cancers, which drastically worsens prognosis for such individuals. Why some SSc patients develop cancer is poorly understood. Clues to this perplexing phenomenon might lie in the inherent genome instability associated with SSc samples. It is well known that cancer is preceded by years of systemic DNA damage and an increase in somatic mutations (i.e. newly acquired, cell specific mutations). Prior studies provide ample evidence of DNA damage in SSc, such as autoimmune responses to key chromatin-associated proteins, cGAS-STING activation, oxidative DNA damage, as well as telomere attrition. In theory, each of these processes can lead to a genome-wide accumulation of deleterious mutations. The discovery and analysis of disease-specific mutations could serve as an ideal readout for prior and ongoing disease associated genotoxic events, and shed light on key mechanisms driving disease progression, and potentially, later cancer development. Indeed, in the last decade, such mutation “signatures” have been described for a variety of DNA damage sources, and have greatly expanded our understanding of how tumors develop.
In our manuscript, we explore the burden of somatic mutagenesis and the prevalent mutation signatures in systemic sclerosis. The key rationale behind our study was that heightened inflammation and immune responses in SSc tissues put genomes under a constant stress, which might drive genome wide mutagenesis. Over time, selection pressures would enrich mutations that allow for cells to escape immune surveillance and/or promote better growth and division, thereby creating ideal conditions for tumorigenesis. We chose lung-derived fibroblasts for our study, primarily for two reasons: 1) Fibroblasts are an ideal cell-type to study a fibrotic disease such as SSc, and are well suited to ex vivo propagation compared to other tissue types, 2) SSc is associated with significant pulmonary complications, and additionally, lung cancers are the most prevalent cancer type observed in SSc patients. Therefore, lung-associated cell types would be likely to accumulate considerable DNA damage and/or mutations. For comparison, we additionally obtained samples from healthy lungs of non-SSc individuals to assess somatic mutation burdens in normal cells. We then asked, do SSc samples carry a heavier mutation burden compared to normal cells, and if so, what can that tell us about the underlying disease mechanism(s)?
We clonally expanded single cells from healthy and SSc lung samples and obtained genomic DNA, on which we performed deep whole genome sequencing. We additionally obtained bulk DNA samples from the initial population of cells, which were subsequently used to estimate the baseline mutations. In this manner, we compared mutation loads across 5 healthy and 9 SSc samples. We then utilized a high-accuracy mutational calling approach that relied on consensus data from multiple independent callers and stringent filters to avoid picking up sub-clonal mutations. Our approach thus gave us the opportunity to identify rare mutational events which would have otherwise been lost amongst background mutations.
Our mutational analysis revealed a remarkable enrichment of single nucleotide variants among SSc samples compared to healthy individuals. Importantly, SSc samples uniquely harbored mutations within known cancer driver genes, as well as genes involved in inflammation and immune response, which suggests that some mutations might be under purifying selection within SSc genomes. We further noticed an enrichment of various other mutational events in individual SSc samples (but rarely or never in healthy samples), including small insertions and deletions (InDels), loss-of-heterozygosity (LOH), copy number variation (CNV), and large-scale variation (including chromosome amplifications/inversions/translocations). Additionally, a subset of SSc samples also displayed a remarkable phenomenon known as “kategis”, or mutation clustering, whereby multiple single nucleotide variants occur within close proximity of each other. Typically, such events are only associated with heavily mutated genomes such as those seen in cancers, and are relatively rare in non-cancer tissues. Our analyses indicate that genomes from SSc patients likely experienced a significant degree of damage, which then manifested as increased downstream mutagenesis.
How did these mutations come about? As mentioned earlier, analyzing the sequence context in the immediate vicinity of mutated bases can reveal “mutation signatures”, that are often specific to the genomic insult (for example CCTT changes upon exposure of DNA to ultraviolet radiation). When SSc-specific mutations were probed against a catalog of somatic mutations, we noticed an enrichment of Signature 93 (SBS93), which predominantly consists of mutations in the trinucleotide context nTw or nCw (where n indicates any DNA base A,T,G, or C, and w indicates an A or T). While there is no previously described etiology for this signature, it appeared remarkably similar to previously-described mutations made by the translesion DNA polymerase Polη (POLH in humans), a specialized DNA polymerase that allows DNA lesion bypass in an error-free manner. We independently verified this using an in-house software, and indeed found an enrichment of POLH-associated nTwN and nCwN signatures in SSc samples. In addition, in a subset of SSc samples, we unexpectedly observed a rare signature often only seen in B-cells, that are derived from the activity of the immune factor AID (wrcT signature). Both POLH and the AID-like signatures were further enriched within the mutational clusters observed earlier, which is yet again a phenomenon that is observed frequently in cancer cells.

Overall, our mutational data reveals a diverse mutational landscape within SSc genomes. A combination of sustained inflammation-associated DNA damage and/or replication errors probably make the most significant contributions to SSc-associated mutagenesis, thereby creating favorable conditions for metastasis (Figure 1). We believe our work represents “ground zero” for further research exploring the nexus of inflammation, immune response, and DNA damage as drivers of disease progression and tumorigenesis, as well as identifying future avenues for diagnosis and therapeutic interventions of rare inflammation-associated diseases.
This work was supported by the National Scleroderma Foundation Research Grant Award to Natalie Saini
----------------
Sriram Vijayraghavan, Ph.D.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in