Navigating prostate cancer progression in humans
Published in Cancer, Chemistry, and Protocols & Methods

The Question
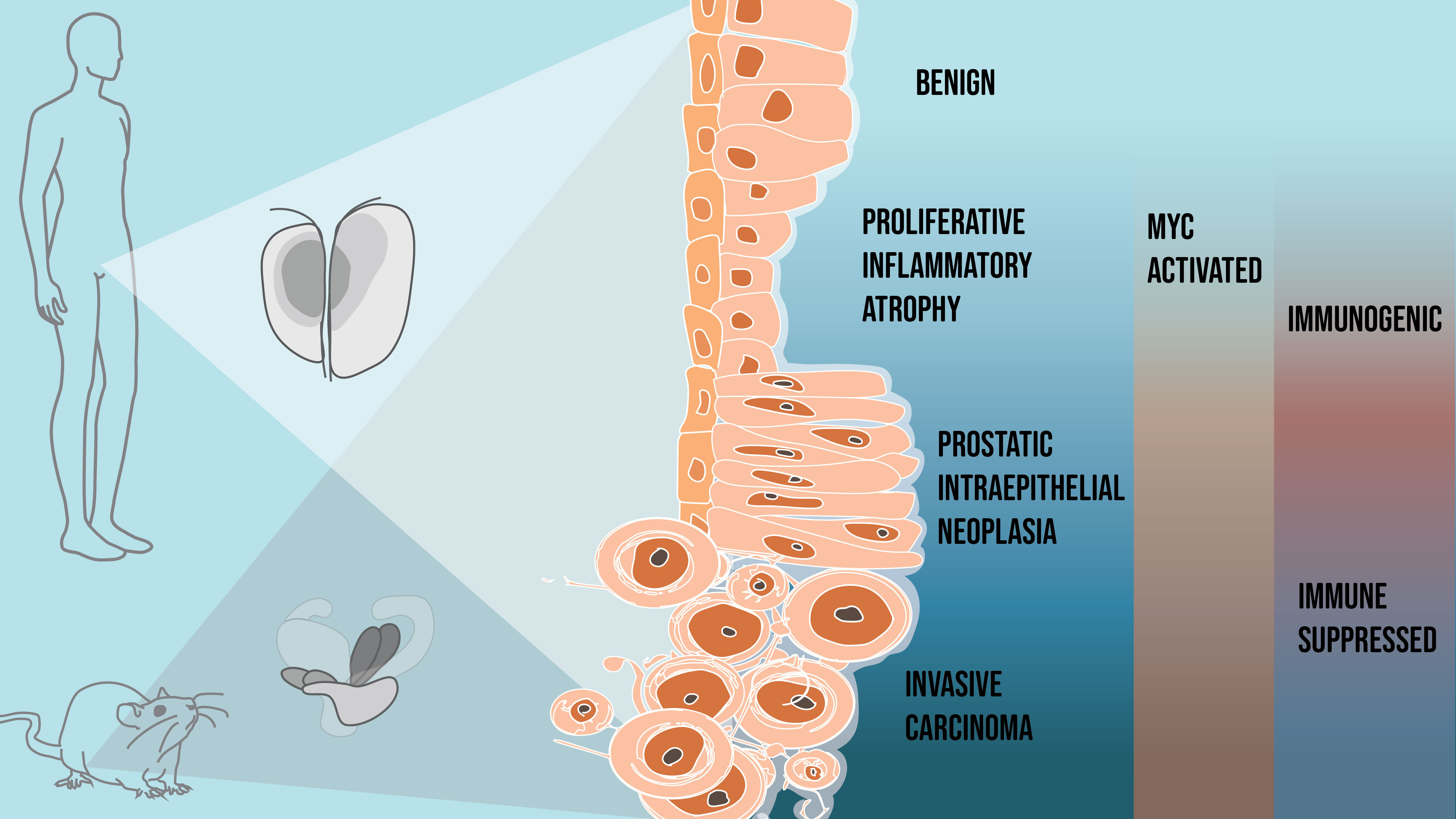
Like many cancers, prostate cancer is a heterogeneous disease, meaning the molecular alterations driving tumorigenesis can be different between patients and even within the same patient. On top of that, the biology of prostate cancer is complex and can be challenging to study. One thing that stuck out in our minds is that despite all these molecular differences, prostate cancer follows a consistent pattern of progression from precursor to invasive disease. You could think of it as a funnel, where the top open end is vast, but it all converges to the same place and direction. We asked the question, why is that? And how can we informatively study this phenomenon with existing tools and technologies?
The Strategy
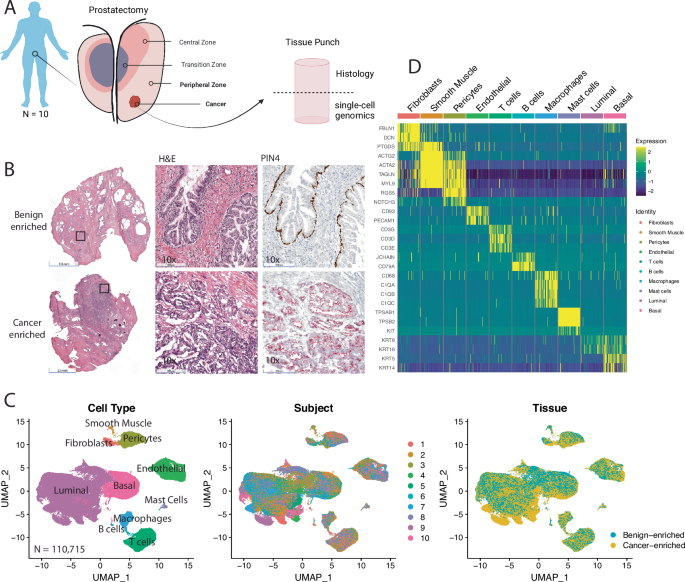
To answer these questions, we used state-of-the-art single-cell RNA sequencing and molecular pathology techniques in patient tissue samples and biologically relevant mouse models in our recently published study led by Dr. Srinivasan Yegnasubramanian and a team of researchers including Dr. Mindy Graham, Rulin Wang, Angelo De Marzo, Chuck Bieberich, Shawn Lupold, Ted DeWeese, Bill Nelson, and several others. One of the challenges of using human prostate tissues is that they are snapshots in time and span multiple prostate cancer types at various stages of progression, with benign cells thrown in there as well. We needed a map with well-defined stages and molecular manipulations to contextualize our information-rich human datasets and make meaningful interpretations. Our strategy was to perform comparative biology of human prostate cancer with multiple genetically engineered mouse models at critical stages of progression to help us navigate these converging changes in the tissues of human prostate cancer.
The Findings
- We found that activation of MYC, a master regulator of genes involved in stem cell pluripotency and carcinogenesis, was commonly shared across human prostate cancer. We observed this in our human scRNA-seq data and confirmed it using the publicly available TCGA primary prostate cancer dataset.
- This motivated us to use MYC-driven models of prostate cancer. We initially started with Hi-Myc mice from two strains: the original model developed in FVB by Charles Sawyers’ lab and Hi-Myc backcrossed into C57BL/6 by co-author Brian Simons. We wanted to ensure we could rule out any strain-specific effects in our analysis. To help us distinguish the MYC-driven changes in the mouse prostate tissues, we relied on our single-cell atlas of the mouse prostate by lobe and strain.
- We found that MYC activity in neoplastic cells reprograms the surrounding tissue directly and indirectly during carcinogenesis. This sets off a cascade of convergent changes to tissue composition and epithelial, immune, and stromal cell states. More importantly, these changes we observed in mouse models could also be seen in our human prostate samples.
- What struck us as we performed our studies was that these changes in the prostate tissue were dynamic. Prostate cancer is notorious for being “immunologically cold”, and we would expect to see tissue changes consistent with something immunosuppressive. However, MYC activation initiated a pro-inflammatory state, paralleling what we see in human proliferative inflammatory atrophy (PIA). PIA was first described and reported by co-authors Angelo De Marzo and Bill Nelson and has been proposed to be a hotbed from which neoplastic prostate epithelial cells can arise.
- Interestingly, we found epithelial populations enriched in the tumor models that did not express the MYC transgene and transcriptionally resembled something between terminally differentiated luminal and basal cells. We referred to these populations as “reactive basal” and “reactive luminal” cells, which have parallels with cells in PIA lesions observed in the human prostate.
- Once the precursor lesions transitioned to invasive carcinoma, we saw the infiltration of regulatory T cells, myeloid-derived suppressor cells, and tumor-associated macrophages characteristic of an immunosuppressive tissue microenvironment. This appeared to be a stark “immunological switch” from the immunogenic signals first appearing with MYC activation in the precursor states to an immunosuppressive state accompanying the transition to invasive cancer.
- Coinciding with these changes in the immune population, we also saw a dramatic change in the stromal population, particularly fibroblasts. One of the key transcriptional changes we saw in these cancer-associated fibroblasts was an elevated expression of genes encoding extracellular matrix proteins, which are consistent with a reactive stroma and the resulting desmoplastic response often described in prostate cancer. In fact, when patients are screened for prostate cancer, clinicians perform digital rectal exams. In these examinations, clinicians feel for a stiffening of the prostate, which happens when there are changes to the composition of the extracellular matrix.
- When MYC-activated neoplastic cells switch from precursor to invasive carcinoma, we found a downregulation of multiple pathways that act as gatekeepers of malignant transformation, including P53 and apoptosis, and upregulation of tumor-promoting pathways such as mTOR signaling. Loss of PTEN, a well-established negative regulator of mTOR signaling, is a frequent alteration in prostate cancer. We hypothesized that losing one of these tumor suppressor pathways would accelerate these MYC-driven tissue changes. To test this, we used the BMPC mouse model that combines MYC activation with PTEN loss, developed by co-authors Chuck Bieberich and Angelo De Marzo. These studies (figures of results found in supplemental information) confirmed our hypothesis and gave us additional insights into the tumor microenvironment of metastatic lesions.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in