

Background
Acute myeloid leukemia (AML) is an aggressive blood cancer caused by the acquisition of heterogenous genetic mutations in hematopoietic stem and progenitor cells. Chemotherapy remains the standard of care for the majority of AML patients and response to treatment depends on age and molecular profile of the patient. Notably, long-term survival of AML patients remains less than 50% overall highlighting the need to develop more effective treatment options.
The ability of cancer cells to evade destruction by the body’s immune system is one of the Hallmarks of Cancer1. Indeed, the discovery that the immune system plays a critical role in controlling the growth of cancer cells has led to revolutionary new treatments known as immunotherapies. Certain types of immunotherapies, including immune checkpoint blockade, have dramatically increased survival of patients with some solid tumour types including melanoma 2. However, these immunotherapies have not been as effective in treating AML, with patients showing only modest responses in clinical trials3. Nevertheless, there is growing evidence to support functional interactions of the immune system with AML cells4. The success of some immunotherapies in the treatment of solid tumours, in particular melanoma, has been attributed to their comparatively greater mutational burden and how this relates to the expression of immunogenic neoantigens that can activate patients' anti-cancer immune response 5,6. In contrast, AML has a relatively low mutational burden and is predicted to express a low number of neoantigens 7,8. This raises the question, what is driving the interaction between the immune system and AML cells and can these interactions be better harnessed in the treatment of patients?
How did we investigate this?
To answer these questions, we investigated the immunogenicity (ability to activate an immune response) of genetically distinct mouse models of AML representing common clinical and prognostic subsets of genetic alterations found in AML patients. The models of AML were generated using overexpression of oncogenes including NUP98-HOX9 + BCR-ABL, MLL-AF9 and AML1-ETO + NrasG12D in mouse hematopoietic stem and progenitor cells. The AMLs were propagated in immunodeficient (Rag2-/-gc-/-) and immunocompetent mice (WT) to investigate immune control. Unexpectedly, we observed loss of the AML1-ETO oncogene in subsequent passages in mice of the AML, leaving only NrasG12D to propagate the disease.
Key findings
We found that AML driven by NrasG12D generated the most immunogenic leukemia cells compared to the other models. Interestingly, we observed that NrasG12D-driven AML expressed the highest level of antigen presentation machinery, MHC Class I and II and CD80 and CD86, the CD28/CTLA-4 ligands (Figure 1). Indeed, human NRAS mutant AML 9 ranked as one of the highest MHC Class II expressing genetic groups.
We found that the immune control of NrasG12D-driven AML was mediated by T cells. NrasG12D AML recipient mice had expanded CD8+ T effector memory cells and displayed a dysfunctional phenotype with, among other exhaustion markers, increased co-expression of PD-1 and TIM-3 (Figure 1). We validated this finding using human single cell RNA-sequencing of AML patients 10 showing that a greater frequency of T cells from mutant RAS AML patients expressed PD-1. We were curious to find out if NrasG12D AML would be sensitive to immune checkpoint blockade by PD-1 inhibition. Indeed, we observed minor but significant effects on AML disease control after PD-1 blockade.
We next asked how NrasG12D-driven AML was able to escape the immune response to propagate disease. We observed increased cell surface expression of immunosuppressive molecules PD-L1 and CD86 on AML cells after exposure to a competent immune system. We also observed selection for AML cells with decreased copy number of NrasG12D integrated into the murine genome. These immunoedited AML cells have decreased Nras gene expression with concurrent upregulation of MYC-driven transcription (Figure 1). We hypothesized that MYC was critical for regulating immune evasion of the AML cells. Indeed, ectopic Myc expression in NrasG12D-driven AML markedly increased PD-L1 and CD86 expression and unexpectedly reduced MHC Class II cell surface expression. Furthermore, NrasG12D + Myc AML had significantly accelerated disease progression compared to NrasG12D AML alone.
Take home message
The question of whether the immune system interacts functionally with AML cells is important to understand if and how to employ immunotherapies for treatment of AML patients. In our study we identify that NrasG12D-driven AML is an immunogenic leukemia that is primarily controlled by T cells. This AML escapes immune control through upregulation of immunosuppressive ligands PD-L1 and CD86, downregulation of NrasG12D and upregulation of MYC-driven transcription (Figure 1). NrasG12D-driven AML responded modestly to PD-1 blockade which may indicate that a subset of AML patients could benefit from treatment with immune checkpoint blockade. However, this would likely be most beneficial in combination with other therapies. Further studies will be required to optimize best use of immunotherapies in AML patients.
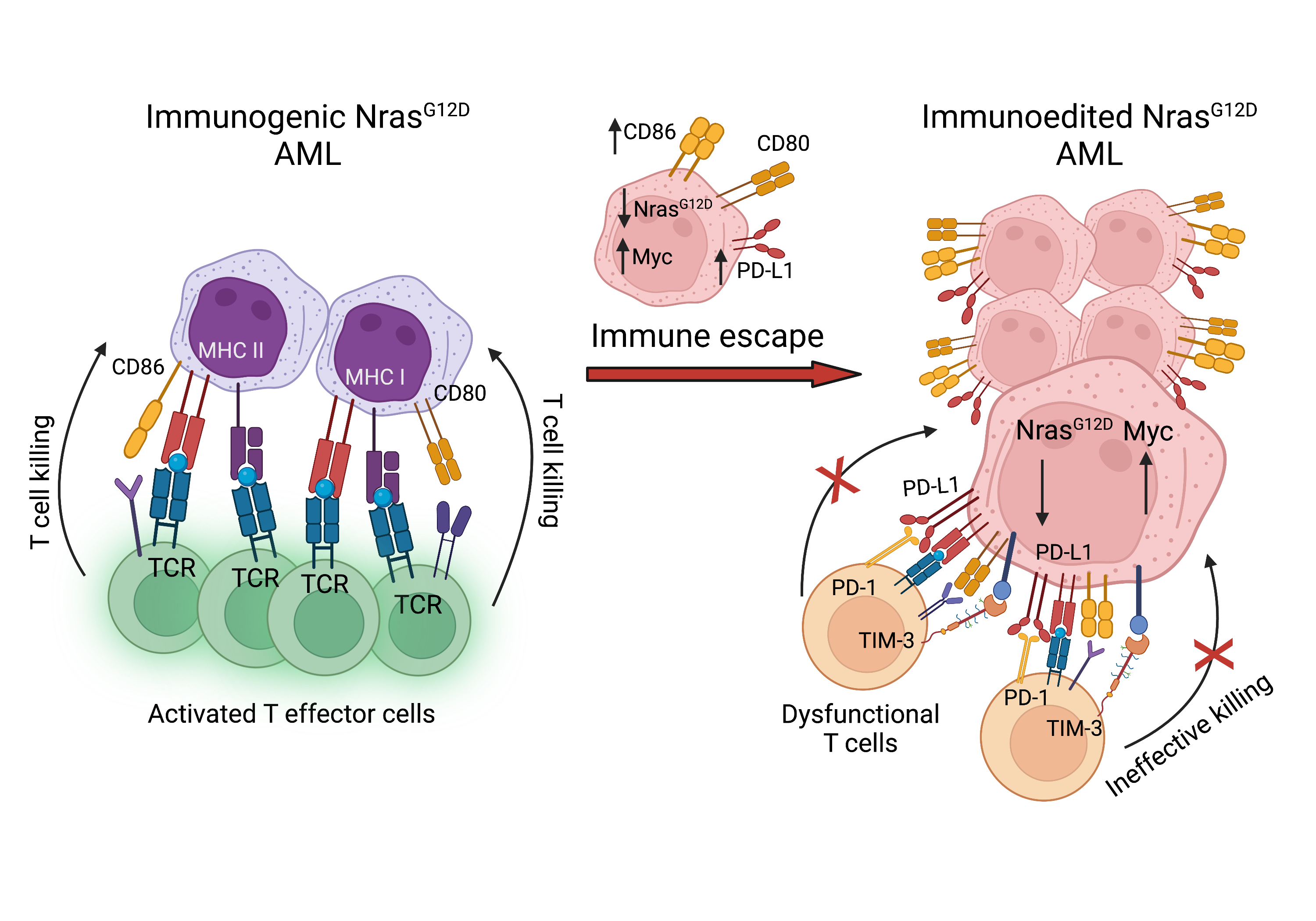
Figure 1: Immune escape of NrasG12D-driven AML. NrasG12D AML cells express high levels of cell surface antigen presentation machinery MHC Class I and II and ligands CD80 and CD86. Immune control of AML cells was mediated by T effector cells. Immune escape of AML cells was facilitated by immunoediting, that is, selection of AML cells with increased cell surface expression of PD-L1 and CD86, decreased expression of NrasG12D and increased MYC-driven transcription. T cells from moribund immunocompetent recipient mice displayed a dysfuntional phenotype with increased co-expression of exhaustion markers including PD-1 and TIM-3. “Created with BioRender.com”.
1 Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov 12, 31-46 (2022). https://doi.org:10.1158/2159-8290.Cd-21-1059
2 Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350-1355 (2018). https://doi.org:10.1126/science.aar4060
3 Liao, D., Wang, M., Liao, Y., Li, J. & Niu, T. A Review of Efficacy and Safety of Checkpoint Inhibitor for the Treatment of Acute Myeloid Leukemia. Front Pharmacol 10, 609 (2019). https://doi.org:10.3389/fphar.2019.00609
4 Austin, R., Smyth, M. J. & Lane, S. W. Harnessing the immune system in acute myeloid leukaemia. Crit Rev Oncol Hematol 103, 62-77 (2016). https://doi.org:10.1016/j.critrevonc.2016.04.020
5 Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Annals of Oncology 30, 44-56 (2019). https://doi.org:https://doi.org/10.1093/annonc/mdy495
6 Hugo, W. et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35-44 (2016). https://doi.org:10.1016/j.cell.2016.02.065
7 Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415-421 (2013). https://doi.org:10.1038/nature12477
8 Rajasagi, M. et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 124, 453-462 (2014). https://doi.org:10.1182/blood-2014-04-567933
9 Verhaak, R. G. et al. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica 94, 131-134 (2009). https://doi.org:10.3324/haematol.13299
10 Lasry, A. et al. An inflammatory state remodels the immune microenvironment and improves risk stratification in acute myeloid leukemia. Nature Cancer 4, 27-42 (2023). https://doi.org:10.1038/s43018-022-00480-0
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026
![When PSMA-targeted therapy is not enough: high-risk localized prostate cancer after repeated [177Lu]Lu-PSMA radioligand therapy](/cdn-cgi/image/metadata=copyright,fit=scale-down,format=auto,quality=95,width=256,height=256/https://public-storage.zapnito.com/Ku6h7Yyp4Q0LXqRRMICCHR2v4LcOsmxMrmDPtOYuI1c)



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in