Phosphoprotein signatures and computational approaches reconstruct patient-specific kinase network topologies
Published in Bioengineering & Biotechnology

Signaling pathways driven by protein and lipid kinases regulate virtually all cell biological functions and are dysregulated in most, if not all, cancers. Current canonical diagrams of cell signaling networks are generic and do not capture how signaling is wired in a specific cell type. This is because our current understanding of kinase networks is an average of data from studies that used different models (fly, mice, human, etc.) and cell types (from different tissues or cancer types). To address this issue, the Cutillas lab has worked for some time in the development of wet-lab and computational methods that can empirically define the topology of a kinase signaling network in a given cell type under defined conditions 1.
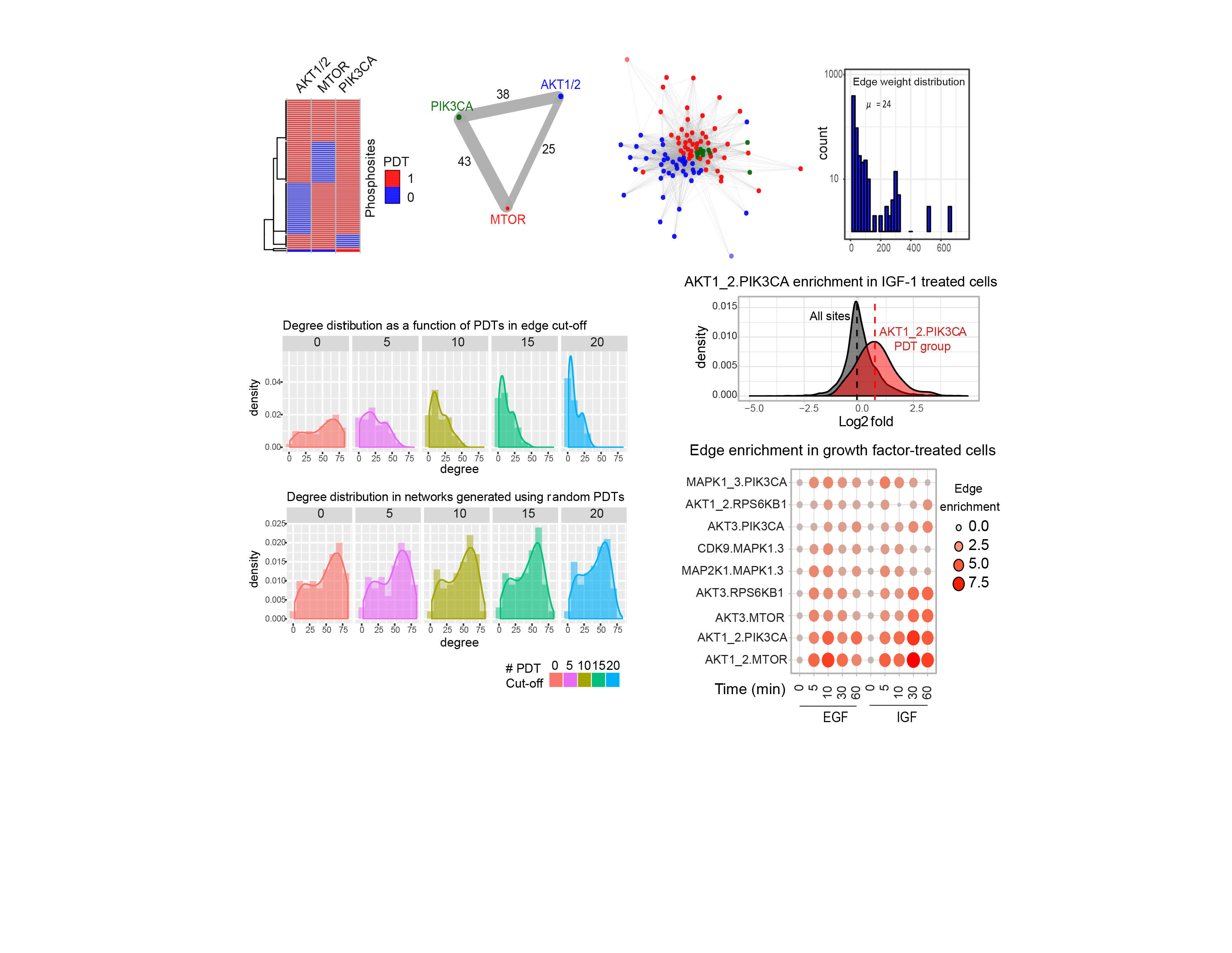
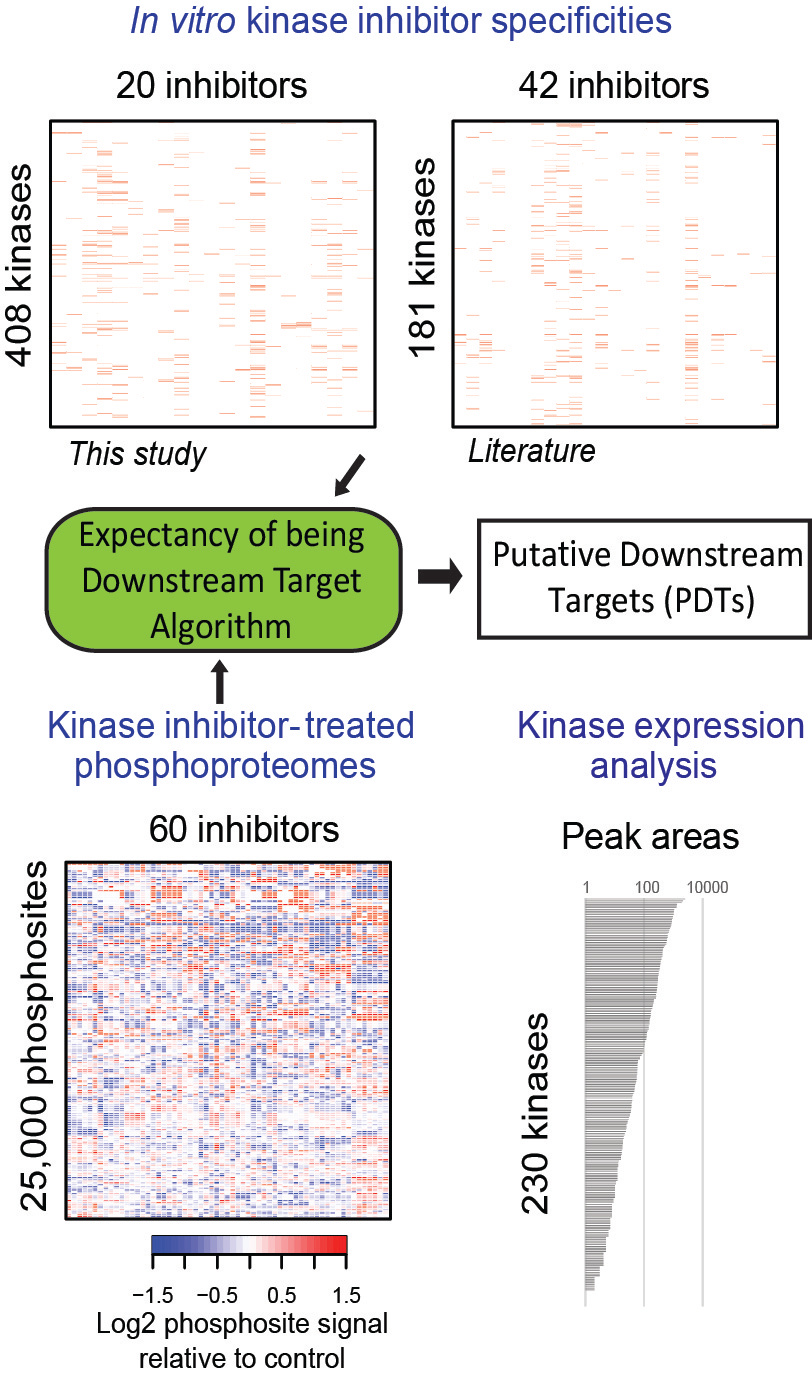
In our recent article in Nature Biotechnology, we aimed to identify kinase network circuitries associated to cancer genotypes and drug sensitivity. To this end, we first designed an algorithm (called “Expectancy of Being Downstream Target” or EBDT) to identify markers of kinase network circuitry (Figure 1). For the approach to work, we first identified sets of small molecule pharmacological compounds that inhibit given kinases. Thus, each kinase is annotated with a kinase inhibitor fingerprint. The EBDT algorithm identifies patterns of phosphorylation that correlate with a kinase’s inhibitor fingerprint. Such correlated sites are regarded as being “Putative Downstream Targets” (or PDTs) for given kinases because we identified >6200 such PDTs for 103 kinases, and validating each site using conventional biochemical techniques would be impossible in practice. PDTs are both direct and indirect kinase substrates; we thus were able to investigate the relationships between kinases and reconstruct a network consisting of 1,508 edges (or kinase-kinase relationships that are conceptualized as signaling axes) between the 103 kinase nodes. In total, we provided substantially new information on downstream targets for kinases poorly annotated in repositories of kinase-substrate relationships.
PDTs are activity markers that are readily quantifiable by mass spectrometry. Using the principles of KSEA 2, we could quantify the enrichment of PDTs and edges across samples. KSEA or “Kinase Substrate Enrichment Analysis” infers kinase activity by calculating enrichment of PDTs for all the kinases in our datasets.
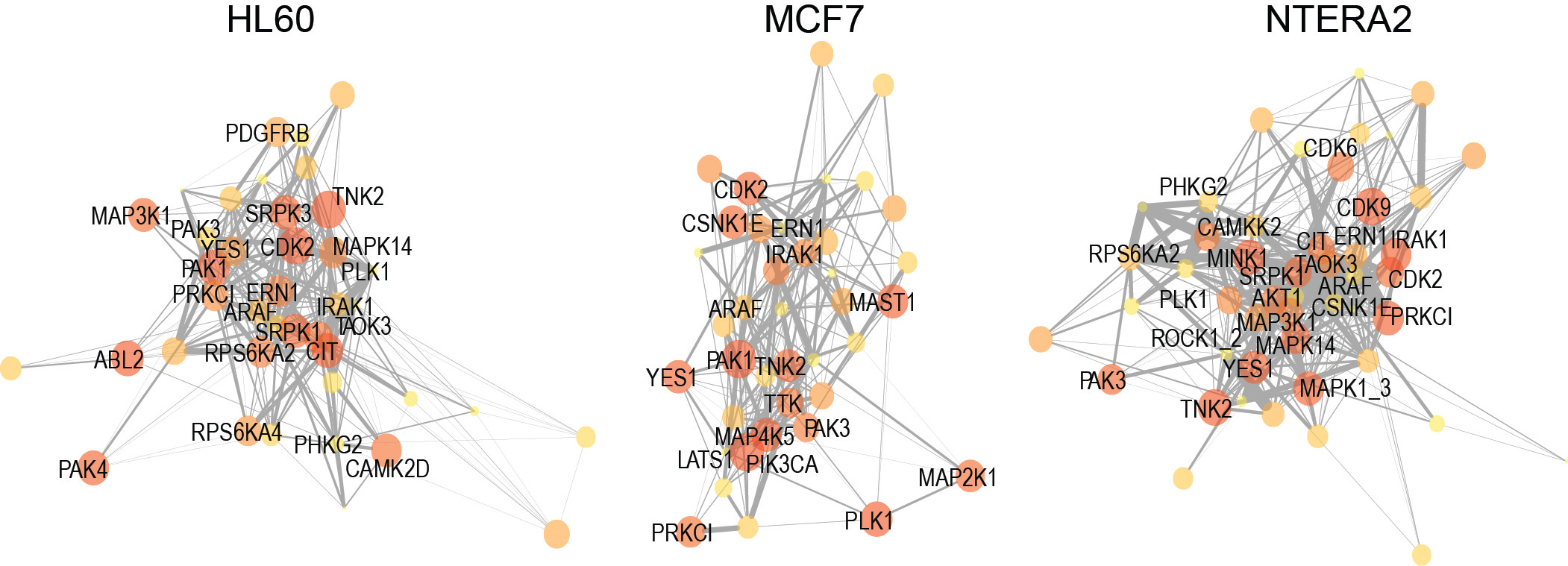
To confirm the biological relevance of our PDTs as markers of kinase network circuitry, we next investigated several of their properties. We found that these groups of phosphorylation sites were enriched in motifs associated to the respective kinase, and behaved as expected in cells treated with agonists and antagonists of kinase signaling. We also used the output from KSEA to reconstruct kinase networks across different cell lines (Figure 2). This methodology, which empirically infers cell-specific kinase networks, allowed us to perform a comparative analysis of topologies across cancer cells of well-defined genetic backgrounds and drug resistant phenotypes. Our work is different from previous methods to analyze kinase signaling, which compare kinase activities or individual phosphorylation sites without providing new information on the kinase-kinase relationships that determine network topology.
To investigate the elements of kinase network topology associated to common genetic mutations, we assessed our markers of circuitry as a function of genetic mutations in primary breast cancer (BC) and acute myeloid leukemia (AML). These markers correlated with the impact that several kinase inhibitors had in reducing the viability of primary AML cases. Multivariate regression models, that used our markers of kinase activity as input, predicted responses to inhibitors with high accuracy. We also found that targeting TTK could be of potential therapeutic value in AML. Finally, we found an anti-correlation between PI3K and MEK-MAPK activation in BC, which we confirmed in cell models isogenic for PIK3CA mutations. Our data suggest that the absence of PIK3CA mutations should be investigated as a predictive signature for MAPK pathway inhibitors and that therapies targeting PI3K signaling in primary breast cancer may have unintended effects in activating MAPK pathways.
In summary, our study provides the cell signaling community with a set of markers of network circuitry and computational approaches to determine topologies associated to the genotypes and drug sensitivity phenotypes of cancer cells.
1. Wilkes, E.H., Terfve, C., Gribben, J.G., Saez-Rodriguez, J. & Cutillas, P.R. Empirical inference of circuitry and plasticity in a kinase signaling network. Proc Natl Acad Sci U S A 112, 7719-7724 (2015).
2. Casado, P. et al. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci Signal 6, rs6 (2013).
Full text link: https://www.nature.com/articles/s41587-019-0391-9
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in